Molecular Phenotypes Segregate Missense Mutations in SLC13A5 Epilepsy
IF 4.5
2区 生物学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
The sodium-coupled citrate transporter (NaCT, SLC13A5) mediates citrate uptake across the plasma membrane via an inward Na+ gradient. Mutations in SLC13A5 cause early infantile epileptic encephalopathy type-25 (EIEE25, SLC13A5 Epilepsy) due to impaired citrate uptake in neurons and astrocytes. Despite clinical identification of disease-causing mutations, underlying mechanisms and cures remain elusive. Here we mechanistically classify six frequent SLC13A5 mutations by phenotyping their protein cell surface expression and citrate transport functions. Mutants C50R, T142M, and T227M exhibit impaired citrate transport despite normal expression at the cell surface. In contrast, mutations G219R, S427L, and L488P show low total protein expression levels, absence of mature, glycosylated proteins at the cell surface, retention of the proteins in the endoplasmic reticulum, and diminished transport activity. This mechanistic classification divides SLC13A5 mutants into two groups, Class I (C50R, T142M, and T227M) and Class II (G219R, S427L, and L488P). Importantly, mutants’ mRNA levels resemble wildtype, suggesting post-translational defects. Class II mutations display immature core-glycosylation and shortened half-lives, indicating protein folding defects. Together, these experiments provide a comprehensive understanding of the disease-causing mutation’s defects in SLC13A5 Epilepsy at the biochemical and molecular level and shed light into the trafficking pathway(s) of NaCT. The two classes of mutations will require fundamentally different approaches for treatment to either restore transport function of the mutant protein that is capable of reaching the cell surface (Class I), or therapies that enable the correction of protein folding defects to enable escape to the cell surface where it may restore transport function (Class II).
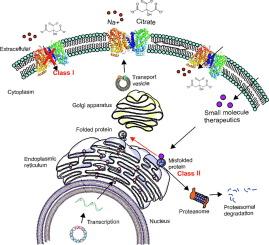
SLC13A5 癫痫错义突变的分子表型分离。
钠偶联柠檬酸盐转运体(NaCT,SLC13A5)通过内向 Na+ 梯度介导柠檬酸盐在质膜上的吸收。由于神经元和星形胶质细胞对柠檬酸盐的摄取能力受损,SLC13A5 基因突变会导致 25 型早期婴儿癫痫性脑病(EIEE25,SLC13A5 Epilepsy)。尽管临床上已发现了致病突变,但其潜在机制和治疗方法仍然难以捉摸。在这里,我们通过表型分析其蛋白细胞表面表达和柠檬酸盐转运功能,从机理上对六种常见的 SLC13A5 突变进行了分类。突变体 C50R、T142M 和 T227M 尽管在细胞表面表达正常,但却表现出柠檬酸盐转运功能受损。相比之下,突变体 G219R、S427L 和 L488P 的蛋白质总表达量较低,细胞表面没有成熟的糖基化蛋白质,蛋白质保留在内质网中,转运活性减弱。这种机理分类法将 SLC13A5 突变体分为两类,一类(C50R、T142M 和 T227M),另一类(G219R、S427L 和 L488P)。重要的是,突变体的 mRNA 水平与野生型相似,表明存在翻译后缺陷。II 类突变体显示出不成熟的核心-糖基化和缩短的半衰期,表明存在蛋白质折叠缺陷。这些实验从生化和分子水平全面了解了致病突变在 SLC13A5 癫痫中的缺陷,并揭示了 NaCT 的贩运途径。这两类突变需要根本不同的治疗方法,要么恢复突变蛋白的转运功能,使其能够到达细胞表面(第一类),要么采用能够纠正蛋白折叠缺陷的疗法,使其能够逃逸到细胞表面,从而恢复转运功能(第二类)。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Molecular Biology
生物-生化与分子生物学
CiteScore
11.30
自引率
1.80%
发文量
412
审稿时长
28 days
期刊介绍:
Journal of Molecular Biology (JMB) provides high quality, comprehensive and broad coverage in all areas of molecular biology. The journal publishes original scientific research papers that provide mechanistic and functional insights and report a significant advance to the field. The journal encourages the submission of multidisciplinary studies that use complementary experimental and computational approaches to address challenging biological questions.
Research areas include but are not limited to: Biomolecular interactions, signaling networks, systems biology; Cell cycle, cell growth, cell differentiation; Cell death, autophagy; Cell signaling and regulation; Chemical biology; Computational biology, in combination with experimental studies; DNA replication, repair, and recombination; Development, regenerative biology, mechanistic and functional studies of stem cells; Epigenetics, chromatin structure and function; Gene expression; Membrane processes, cell surface proteins and cell-cell interactions; Methodological advances, both experimental and theoretical, including databases; Microbiology, virology, and interactions with the host or environment; Microbiota mechanistic and functional studies; Nuclear organization; Post-translational modifications, proteomics; Processing and function of biologically important macromolecules and complexes; Molecular basis of disease; RNA processing, structure and functions of non-coding RNAs, transcription; Sorting, spatiotemporal organization, trafficking; Structural biology; Synthetic biology; Translation, protein folding, chaperones, protein degradation and quality control.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


