Highly Efficient Solar Energy Harvesting in Phosphorene–Graphene Quantum Dot van der Waals Heterostructures: An Ab Initio Approach
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
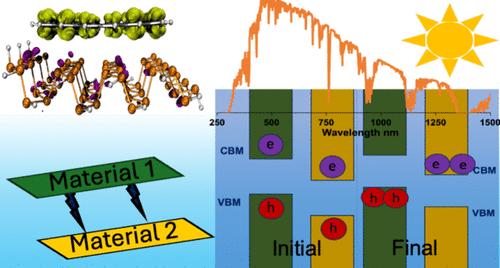
This study delves into the intricacies of creating highly effective power conversion assemblies from van der Waals heterostructures of phosphorene and graphene quantum dots by employing density functional theory calculations. We emphasize the role of individual monomer properties and their interlayer interactions in the power conversion ability by focusing on visible light absorption, charge carrier generation, and their separation. Different edge atom functionalization (H, NH2, Cl, OCN, CN) in the phosphorene quantum dots and heteroatom doping (Group IV = Si, Ge, and Group VI = O, S, Se) in the basal plane of graphene quantum dots were employed to alter the monomer properties. Our results indicate that select combinations of these modification techniques yield staggered frontier molecular orbital alignments (Type II) with spatial separation of the highest occupied and lowest unoccupied molecular orbitals. These candidates possess an improved visible light absorption range with reduced intensities owing to the dominance of charge transfer excitations. Edge functionalization of phosphorene was identified as the most significant contributor to interlayer interaction strength, with functional groups that are electron-withdrawing in nature forming stronger interactions. Heteroatom doping of graphene was recognized as the most important contributor to improving visible light absorbance owing to the reduction in fundamental gaps. From the candidates considered, systems with relatively weaker interlayer interactions were determined to be better at charge carrier separation due to the potential gradient being concentrated at the interfacial region. These systems possess approximated power conversion efficiencies ranging between 11 and 29%, among the highest reported for quantum dot systems characterized by density functional theory calculations.
磷化烯-石墨烯量子点范德华异质结构中的高效太阳能收集:一种 Ab Initio 方法
本研究通过密度泛函理论计算,深入探讨了利用磷化物和石墨烯量子点的范德华异质结构创建高效功率转换组件的复杂性。我们通过关注可见光吸收、电荷载流子生成及其分离,强调了单个单体特性及其层间相互作用在功率转换能力中的作用。我们采用了磷烯量子点中不同的边缘原子官能化(H、NH2、Cl、OCN、CN)和石墨烯量子点基底面中的杂原子掺杂(第四组=Si、Ge,第六组=O、S、Se)来改变单体特性。我们的研究结果表明,这些改性技术的精选组合可产生交错的前沿分子轨道排列(第二类),最高占有和最低未占有分子轨道在空间上相互分离。由于电荷转移激发占主导地位,这些候选物质的可见光吸收范围得到改善,强度降低。研究发现,磷烯的边缘官能化是影响层间相互作用强度的最重要因素,具有电子吸收性质的官能团会形成更强的相互作用。石墨烯的异构体掺杂被认为是提高可见光吸收率的最重要因素,这是因为基本间隙减小了。在考虑的候选系统中,具有相对较弱层间相互作用的系统被认为在电荷载流子分离方面更胜一筹,这是因为电势梯度集中在界面区域。这些系统的近似功率转换效率介于 11% 和 29% 之间,是密度泛函理论计算量子点系统中最高的。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


