Quantitation of metabolic activity from isotope tracing data using automated methodology
IF 18.9
1区 医学
Q1 ENDOCRINOLOGY & METABOLISM
引用次数: 0
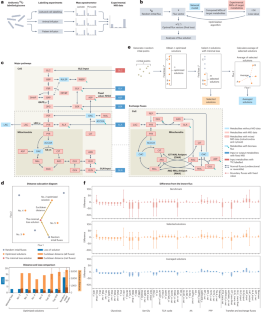

利用自动方法从同位素追踪数据中量化代谢活动
代谢通量分析(MFA)是一种基于机器学习原理破译标记模式的计算方法。与典型的机器学习算法从已知数据集训练模型进行预测不同,常用的 MFA 算法利用同位素追踪实验的数据训练代谢网络,并直接输出学习到的信息,即网络中最适合数据的所有通量3,5(图 1b)。然而,作为一种机器学习算法,目前的 MFA 方法往往缺乏系统的评估和基准测试,而这是更广泛的机器学习和人工智能应用中的标准做法6。MFA 研究中的算法收敛性、通量估计准确性和结果稳健性等问题已被提出,但在很大程度上仍未得到解决3,这限制了这些自动化工具在代谢研究中的有效性和广泛应用。该模型包含关键通路中的 100 多个通量,包括糖酵解、三羧酸(TCA)循环、磷酸戊糖通路(PPP)、一碳代谢和几个氨基酸(AA)生物合成通路(图 1c,补充方法)。与当代的 MFA 工具7,8,9 相比,我们的方法的一个显著特点是纳入了细胞器分区,有助于准确量化真核细胞线粒体和细胞膜之间的交换通量(图 1c)。其他工具通常需要几十分钟才能得到一个解决方案7,9,而我们的方法可以在台式电脑上10 在 2 秒钟内生成一个优化解决方案,其通量可以准确解释培养细胞系 13C 追踪实验的标记模式(补充图 1a-d)。为了解决这个问题,我们开发了一种优化-平均算法,该算法通过从优化解决方案池中选择损失最小的解决方案子集(选定解决方案),并对其进行平均,从而生成一个新的、更稳定的解决方案集(平均解决方案)(图 1e,补充方法)来完善计算过程。使用已知通量矢量生成的模拟 13C 追踪数据集(补充图 2a)对这些解决方案以及使用当代软件中使用的典型策略生成的解决方案(补充方法,补充图 1c)进行了基准测试。结果表明,与基准相比,优化平均算法有效地减少了通量的变化,并提高了近似已知通量结果的准确性,即使数据可用性水平不同也是如此(图 1f,补充图 2b-e 和 3a-e)。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature metabolism
ENDOCRINOLOGY & METABOLISM-
CiteScore
27.50
自引率
2.40%
发文量
170
期刊介绍:
Nature Metabolism is a peer-reviewed scientific journal that covers a broad range of topics in metabolism research. It aims to advance the understanding of metabolic and homeostatic processes at a cellular and physiological level. The journal publishes research from various fields, including fundamental cell biology, basic biomedical and translational research, and integrative physiology. It focuses on how cellular metabolism affects cellular function, the physiology and homeostasis of organs and tissues, and the regulation of organismal energy homeostasis. It also investigates the molecular pathophysiology of metabolic diseases such as diabetes and obesity, as well as their treatment. Nature Metabolism follows the standards of other Nature-branded journals, with a dedicated team of professional editors, rigorous peer-review process, high standards of copy-editing and production, swift publication, and editorial independence. The journal has a high impact factor, has a certain influence in the international area, and is deeply concerned and cited by the majority of scholars.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


