Distinct roles for the domains of the mitochondrial aspartate/glutamate carrier citrin in organellar localization and substrate transport
IF 7
2区 医学
Q1 ENDOCRINOLOGY & METABOLISM
引用次数: 0
Abstract
Objective
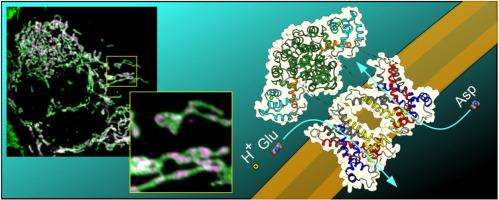
Citrin, the mitochondrial aspartate/glutamate carrier isoform 2 (AGC2), is structurally and mechanistically the most complex SLC25 family member, because it consists of three domains and forms a homo-dimer. Each protomer has an N-terminal calcium-binding domain with EF-hands, followed by a substrate-transporting carrier domain and a C-terminal domain with an amphipathic helix. The absence or dysfunction of citrin leads to citrin deficiency, a highly prevalent pan-ethnic mitochondrial disease. Here, we aim to understand the role of different citrin domains and how they contribute to pathogenic mechanisms in citrin deficiency.
Methods
We have employed structural modeling and functional reconstitution of purified proteins in proteoliposomes to assess the transport activity and calcium regulation of wild-type citrin and pathogenic variants associated with citrin deficiency. We have also developed a double knockout of citrin and aralar (AGC1), the two paralogs of the mitochondrial aspartate/glutamate carrier, in HAP1 cells to perform mitochondrial imaging and to investigate mitochondrial localisation.
Results
Using 33 pathogenic variants of citrin we clarify determinants of subcellular localization and transport mechanism. We identify crucial elements of the carrier domain that are required for transport, including those involved in substrate binding, network formation and dynamics. We show that the N-terminal domain is not involved in calcium regulation of transport, as previously thought, but when mutated causes a mitochondrial import defect.
Conclusions
Our work introduces a new role for the N-terminal domain of citrin and demonstrates that dysfunction of the different domains contributes to distinct pathogenic mechanisms in citrin deficiency.
线粒体天冬氨酸/谷氨酸载体柠檬蛋白的结构域在细胞器定位和底物运输中的不同作用
目的:线粒体天冬氨酸/谷氨酸载体同工酶 2 是结构和机理上最复杂的 SLC25 家族成员,因为它由三个结构域组成,并形成一个同源二聚体。每个原体都有一个带有 EF 手的 N 端钙结合结构域、一个底物运输载体结构域和一个带有两性螺旋的 C 端结构域。柠檬蛋白的缺失或功能障碍导致柠檬蛋白缺乏症,这是一种高发的泛种族线粒体疾病。在此,我们旨在了解不同 citrin 结构域的作用,以及它们如何在 citrin 缺乏症的致病机制中发挥作用:方法:我们利用结构建模和蛋白脂质体中纯化蛋白的功能重组来评估野生型citrin和与citrin缺乏症相关的致病变体的转运活性和钙调控。我们还在HAP1细胞中开发了线粒体天冬氨酸/谷氨酸载体的两个旁系亲属--citrin和aralar(AGC1)的双基因敲除,以进行线粒体成像并研究线粒体定位:结果:我们利用柠檬蛋白的 33 个致病变体阐明了亚细胞定位和转运机制的决定因素。我们确定了运输所需的载体结构域的关键元素,包括参与底物结合、网络形成和动力学的元素。我们发现 N 端结构域并不像以前认为的那样参与运输的钙调控,而是在发生突变时导致线粒体输入缺陷:我们的研究为柠檬蛋白的 N 端结构域引入了一个新的角色,并证明不同结构域的功能障碍会导致柠檬蛋白缺乏症的不同致病机制。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Molecular Metabolism
ENDOCRINOLOGY & METABOLISM-
CiteScore
14.50
自引率
2.50%
发文量
219
审稿时长
43 days
期刊介绍:
Molecular Metabolism is a leading journal dedicated to sharing groundbreaking discoveries in the field of energy homeostasis and the underlying factors of metabolic disorders. These disorders include obesity, diabetes, cardiovascular disease, and cancer. Our journal focuses on publishing research driven by hypotheses and conducted to the highest standards, aiming to provide a mechanistic understanding of energy homeostasis-related behavior, physiology, and dysfunction.
We promote interdisciplinary science, covering a broad range of approaches from molecules to humans throughout the lifespan. Our goal is to contribute to transformative research in metabolism, which has the potential to revolutionize the field. By enabling progress in the prognosis, prevention, and ultimately the cure of metabolic disorders and their long-term complications, our journal seeks to better the future of health and well-being.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


