{"title":"Pre-training strategy for antiviral drug screening with low-data graph neural network: A case study in HIV-1 K103N reverse transcriptase.","authors":"Kajjana Boonpalit, Hathaichanok Chuntakaruk, Jiramet Kinchagawat, Peter Wolschann, Supot Hannongbua, Thanyada Rungrotmongkol, Sarana Nutanong","doi":"10.1002/jcc.27514","DOIUrl":null,"url":null,"abstract":"<p><p>Graph neural networks (GNN) offer an alternative approach to boost the screening effectiveness in drug discovery. However, their efficacy is often hindered by limited datasets. To address this limitation, we introduced a robust GNN training framework, applied to various chemical databases to identify potent non-nucleoside reverse transcriptase inhibitors (NNRTIs) against the challenging K103N-mutated HIV-1 RT. Leveraging self-supervised learning (SSL) pre-training to tackle data scarcity, we screened 1,824,367 compounds, using multi-step approach that incorporated machine learning (ML)-based screening, analysis of absorption, distribution, metabolism, and excretion (ADME) prediction, drug-likeness properties, and molecular docking. Ultimately, 45 compounds were left as potential candidates with 17 of the compounds were previously identified as NNRTIs, exemplifying the model's efficacy. The remaining 28 compounds are anticipated to be repurposed for new uses. Molecular dynamics (MD) simulations on repurposed candidates unveiled two promising preclinical drugs: one designed against Plasmodium falciparum and the other serving as an antibacterial agent. Both have superior binding affinity compared to anti-HIV drugs. This conceptual framework could be adapted for other disease-specific therapeutics, facilitating the identification of potent compounds effective against both WT and mutants while revealing novel scaffolds for drug design and discovery.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":" ","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1002/jcc.27514","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
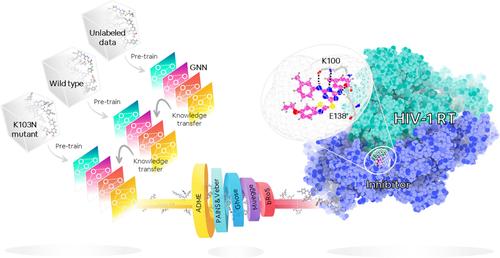
Abstract
Graph neural networks (GNN) offer an alternative approach to boost the screening effectiveness in drug discovery. However, their efficacy is often hindered by limited datasets. To address this limitation, we introduced a robust GNN training framework, applied to various chemical databases to identify potent non-nucleoside reverse transcriptase inhibitors (NNRTIs) against the challenging K103N-mutated HIV-1 RT. Leveraging self-supervised learning (SSL) pre-training to tackle data scarcity, we screened 1,824,367 compounds, using multi-step approach that incorporated machine learning (ML)-based screening, analysis of absorption, distribution, metabolism, and excretion (ADME) prediction, drug-likeness properties, and molecular docking. Ultimately, 45 compounds were left as potential candidates with 17 of the compounds were previously identified as NNRTIs, exemplifying the model's efficacy. The remaining 28 compounds are anticipated to be repurposed for new uses. Molecular dynamics (MD) simulations on repurposed candidates unveiled two promising preclinical drugs: one designed against Plasmodium falciparum and the other serving as an antibacterial agent. Both have superior binding affinity compared to anti-HIV drugs. This conceptual framework could be adapted for other disease-specific therapeutics, facilitating the identification of potent compounds effective against both WT and mutants while revealing novel scaffolds for drug design and discovery.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


