NLOphoric unsymmetrically substituted D-π-A benzodifuranone dyes: Density functional theory, time dependent-density functional theory, and non-linear optical studies
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
The linear and non-linear optical properties of thirty-five unsymmetrically substituted compounds are studied using density functional theory and time-dependent density functional theory methods (B3LYP/6311++G(d,p), CAM-B3LYP/6311++G(d,p) and M06-2X/6311++G(d,p)). A comparable set of functionals is also used to investigate vertical excitation. It is observed that the inclusion of the nitrogen-substituted electron-donating group along with the electron-withdrawing group led to more red-shifted absorption maxima and exhibited an excellent (non-linear optical) NLO response. The geometrical framework, dipole moment, and other descriptors, HOMO-LUMO energy gaps, linear polarizability, first-order and second-order hyperpolarizability are calculated to investigate the effect of different electron donating and accepting substituents on the NLO properties of benzodifuranone chromophores. In cases where nitrogen substituents are added, the electron density is more widely spread throughout the donor in HOMO and more displaced to the acceptor in LUMO. The computed first-order and second-order hyperpolarizability values and decreasing HOMO-LUMO energy gaps in disubstituted nitrogen-containing compounds show that 1a-1e, 4a-4e, 5a-5e are promising candidates in all functionals for better NLO properties.
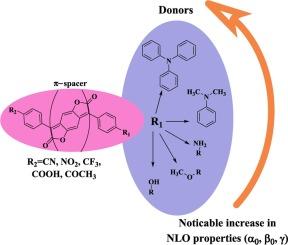
非对称取代 D-π-A 苯并二呋喃酮染料的非线性光学:密度泛函理论、时变密度泛函理论和非线性光学研究
采用密度泛函理论和时变密度泛函理论方法(B3LYP/6311++G(d,p)、CAM-B3LYP/6311++G(d,p) 和 M06-2X/6311++G(d,p))研究了 35 种不对称取代化合物的线性和非线性光学性质。一组类似的函数也用于研究垂直激发。研究发现,氮取代的电子供体基团和电子吸收基团的加入导致了更多的红移吸收最大值,并表现出优异的(非线性光学)NLO 响应。通过计算几何框架、偶极矩和其他描述符、HOMO-LUMO 能隙、线性极化率、一阶和二阶超极化率,研究了不同的供电子和受电子取代基对苯并二呋喃酮发色团 NLO 特性的影响。在加入氮取代基的情况下,电子密度在 HOMO 中更广泛地分布于整个供体,而在 LUMO 中则更多地转移到受体。计算得出的含氮二取代化合物的一阶和二阶超极化率值以及不断减小的 HOMO-LUMO 能隙表明,1a-1e、4a-4e 和 5a-5e 在所有函数中都有希望获得更好的 NLO 性能。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


