A diagnosis of non-neuronopathic and late-onset acid sphingomyelinase deficiency (Niemann-Pick disease A/B) following bone marrow biopsy showing foamy histiocytosis
Ivo N. SahBandar, Gustavo H. B. Maegawa, Danielle Brandman, Jacob H. Rand, Hana I. Lim, Julia T. Geyer
{"title":"A diagnosis of non-neuronopathic and late-onset acid sphingomyelinase deficiency (Niemann-Pick disease A/B) following bone marrow biopsy showing foamy histiocytosis","authors":"Ivo N. SahBandar, Gustavo H. B. Maegawa, Danielle Brandman, Jacob H. Rand, Hana I. Lim, Julia T. Geyer","doi":"10.1002/jha2.1003","DOIUrl":null,"url":null,"abstract":"<p>The patient was a 44-year-old East-Asian descent male presenting with longstanding splenomegaly, thrombocytopenia, easy bruising since childhood, and cryptogenic cirrhosis with a history of recurrent variceal bleeding, currently being evaluated for a liver transplant. Pertinent laboratory findings include low serum albumin (3.6, <i>N</i> = 3.9–5.2 g/dL), increased total (4.8, <i>N</i> = 0.3–1.2 mg/dL), direct (1.3, <i>N</i> = ≤ 0.3 mg/dL), and indirect bilirubin (3.5, <i>N</i> = 0.1–0.8 mg/dL), mild AST elevation (53, <i>N</i> = ≤ 34 U/L), mild normocytic anemia (Hgb 119, <i>N</i> = 126–170 g/L, MCV 85.9, <i>N</i> = 78.6–94.2 fL), and thrombocytopenia (22 × 10e9, <i>N</i> = 156–325 × 10e9/L).</p><p>The bone marrow biopsy and clot section showed erythroid hyperplasia, decreased granulopoiesis with complete maturation for both lineages, and reduced megakaryocytes (Figure 1, panel A, H&E, 40X original magnification). A significant number of foamy histiocytes were identified, some of which contained cytoplasmic red blood cells (erythrophagocytosis, panel A). While most macrophages were negative for Periodic acid–Schiff (PAS) special stain, rare PAS-positive macrophages were seen (black arrow; Figure 1, panel B, PAS special stain, 40X original magnification). The described findings ruled out the presence of glycogen storage or Whipple disease. Representative macrophages (Figure 1, panels C–E, Wright-Giemsa, 100X original magnification) show typical features of acid sphingomyelinase deficiency (ASMD, aka Niemann-Pick disease types A/B) with low nuclear to cytoplasmic ratio and ample uniformly finely vacuolated cytoplasm, some contained red blood cells and debris (panel D), and sea blue histiocytes with deeply basophilic cytoplasm were noted (panel E). No organisms were identified by Grocott-Gomori methenamine silver (GMS) staining, and immunohistochemistry studies showed foamy histocytes non-reactivity to S100, langerin, and BRAF (figures not shown).</p><p>The clinical history and bone marrow biopsy findings were suspicious for lysosomal storage disease, and subsequent sphingomyelinase enzymatic activity (dried-blot spot, DBS) showed decreased residual activity (0.4 nmol/L, N ≥ 2.5 nmol/L) consistent with the late-onset non-neuronopathic form of ASMD, which is characterized by the development of hepatosplenomegaly and associated thrombocytopenia. In addition, the oxysterol, cholestane-3beta,5alpha- 6beta-triol (1.0 nmol/mL, <i>N</i> ≤ 0.8), and lyso-sphingomyelin (0.582 nmol/mL <i>N</i> ≤ 0.100) were elevated in DBS. Interestingly, chitotriosidase (492 nmols/h/mL, <i>N</i> 4–120) and angiotensin-converting enzyme (114 IU/L, <i>N</i> 16–85) were also elevated, reflecting the expanded reticulum endothelial system. Other lysosomal enzymes were at normal levels, as well as other sphingolipids, including lyso-glucosylphingosine. The case illustrates the importance of identifying bone marrow lipid-laden foam cells, triggering investigations for ASMD, to which Food and Drug Administration-approved disease-modifying therapy olipudase alpha (Xenpozyme), which can significantly improve the patient's splenomegaly, is currently available.</p><p>Ivo N. SahBandar and Julia T. Geyer designed the study, Ivo N. SahBandar, Julia T. Geyer, Gustavo H. B. Maegawa, and Jacob H. Rand conceived and analyzed the clinical and histologic data, Danielle Brandman and Hana I. Lim provided clinical data, Ivo N. SahBandar and Julia T. Geyer prepared the manuscript, Ivo N. SahBandar, Julia T. Geyer, Gustavo H. B. Maegawa, Hana I. Lim, Danielle Brandman and Jacob H. Rand edited the manuscript.</p><p>The authors declare no conflict of interest.</p><p>The authors received no specific funding for this work.</p><p>This study protocol was reviewed and approved by the Weill Cornell Medicine Institutional Review Board (WCM-IRB) at Weill Cornell Medical College of Cornell University, approval number 0107004999.</p><p>Written informed consent was obtained from the patient for publication of the details of their medical case and any accompanying images. Only deidentified data were used in this manuscript, and no information revealing the patient's identity was included. The New York Presbyterian Hospital/Weill Cornell Medicine is in compliance with the CARE guidelines for case reports.</p><p>The authors have confirmed clinical trial registration is not needed for this submission.</p>","PeriodicalId":72883,"journal":{"name":"EJHaem","volume":"5 5","pages":"1078-1079"},"PeriodicalIF":0.0000,"publicationDate":"2024-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jha2.1003","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"EJHaem","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jha2.1003","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
The patient was a 44-year-old East-Asian descent male presenting with longstanding splenomegaly, thrombocytopenia, easy bruising since childhood, and cryptogenic cirrhosis with a history of recurrent variceal bleeding, currently being evaluated for a liver transplant. Pertinent laboratory findings include low serum albumin (3.6, N = 3.9–5.2 g/dL), increased total (4.8, N = 0.3–1.2 mg/dL), direct (1.3, N = ≤ 0.3 mg/dL), and indirect bilirubin (3.5, N = 0.1–0.8 mg/dL), mild AST elevation (53, N = ≤ 34 U/L), mild normocytic anemia (Hgb 119, N = 126–170 g/L, MCV 85.9, N = 78.6–94.2 fL), and thrombocytopenia (22 × 10e9, N = 156–325 × 10e9/L).
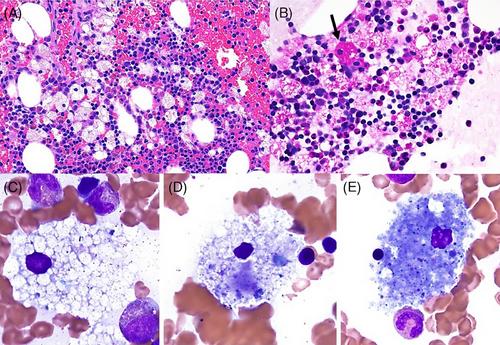
The bone marrow biopsy and clot section showed erythroid hyperplasia, decreased granulopoiesis with complete maturation for both lineages, and reduced megakaryocytes (Figure 1, panel A, H&E, 40X original magnification). A significant number of foamy histiocytes were identified, some of which contained cytoplasmic red blood cells (erythrophagocytosis, panel A). While most macrophages were negative for Periodic acid–Schiff (PAS) special stain, rare PAS-positive macrophages were seen (black arrow; Figure 1, panel B, PAS special stain, 40X original magnification). The described findings ruled out the presence of glycogen storage or Whipple disease. Representative macrophages (Figure 1, panels C–E, Wright-Giemsa, 100X original magnification) show typical features of acid sphingomyelinase deficiency (ASMD, aka Niemann-Pick disease types A/B) with low nuclear to cytoplasmic ratio and ample uniformly finely vacuolated cytoplasm, some contained red blood cells and debris (panel D), and sea blue histiocytes with deeply basophilic cytoplasm were noted (panel E). No organisms were identified by Grocott-Gomori methenamine silver (GMS) staining, and immunohistochemistry studies showed foamy histocytes non-reactivity to S100, langerin, and BRAF (figures not shown).
The clinical history and bone marrow biopsy findings were suspicious for lysosomal storage disease, and subsequent sphingomyelinase enzymatic activity (dried-blot spot, DBS) showed decreased residual activity (0.4 nmol/L, N ≥ 2.5 nmol/L) consistent with the late-onset non-neuronopathic form of ASMD, which is characterized by the development of hepatosplenomegaly and associated thrombocytopenia. In addition, the oxysterol, cholestane-3beta,5alpha- 6beta-triol (1.0 nmol/mL, N ≤ 0.8), and lyso-sphingomyelin (0.582 nmol/mL N ≤ 0.100) were elevated in DBS. Interestingly, chitotriosidase (492 nmols/h/mL, N 4–120) and angiotensin-converting enzyme (114 IU/L, N 16–85) were also elevated, reflecting the expanded reticulum endothelial system. Other lysosomal enzymes were at normal levels, as well as other sphingolipids, including lyso-glucosylphingosine. The case illustrates the importance of identifying bone marrow lipid-laden foam cells, triggering investigations for ASMD, to which Food and Drug Administration-approved disease-modifying therapy olipudase alpha (Xenpozyme), which can significantly improve the patient's splenomegaly, is currently available.
Ivo N. SahBandar and Julia T. Geyer designed the study, Ivo N. SahBandar, Julia T. Geyer, Gustavo H. B. Maegawa, and Jacob H. Rand conceived and analyzed the clinical and histologic data, Danielle Brandman and Hana I. Lim provided clinical data, Ivo N. SahBandar and Julia T. Geyer prepared the manuscript, Ivo N. SahBandar, Julia T. Geyer, Gustavo H. B. Maegawa, Hana I. Lim, Danielle Brandman and Jacob H. Rand edited the manuscript.
The authors declare no conflict of interest.
The authors received no specific funding for this work.
This study protocol was reviewed and approved by the Weill Cornell Medicine Institutional Review Board (WCM-IRB) at Weill Cornell Medical College of Cornell University, approval number 0107004999.
Written informed consent was obtained from the patient for publication of the details of their medical case and any accompanying images. Only deidentified data were used in this manuscript, and no information revealing the patient's identity was included. The New York Presbyterian Hospital/Weill Cornell Medicine is in compliance with the CARE guidelines for case reports.
The authors have confirmed clinical trial registration is not needed for this submission.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


