Local potential energy density – A DFT analysis and the local binding energy in complexes with multiple interactions
IF 2.7
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
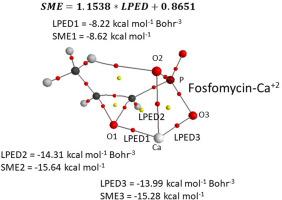
A recently developed method, so-called local potential energy density, LPED, provides the binding energy density of intra/intermolecular interactions. The LPED cannot directly give the binding energy of intra/intermolecular interactions. However, it can indirectly give the binding energy through the linear equation between LPED and supramolecular binding energy, SME. In addition, the LPED can be used to obtain the SME of local or individual interactions indirectly for the case of complexes with multiple interactions, which cannot be obtained for any other method to our knowledge. The calculation of the LPED was evaluated with three different levels of theory using density functional methods. The linearity of LPED and SME between the reference level of theory (ωB97X-D/aug-cc-pVTZ) and the other levels of theory are similar among the studied levels of theory. In addition, LPED was used indirectly to obtain the local binding energy of intermolecular interactions of complexes with multiple interactions, such as the EDTA-Ca+2 and the fosfomycin-Ca+2.
局部势能密度 - DFT 分析和具有多重相互作用的复合物的局部结合能
最近开发的一种方法,即所谓的局部势能密度(LPED),可提供分子内/分子间相互作用的结合能密度。LPED 不能直接给出分子内/分子间相互作用的结合能。不过,它可以通过 LPED 与超分子结合能 SME 之间的线性方程间接给出结合能。此外,对于具有多重相互作用的复合物,LPED 还可用于间接获得局部或单个相互作用的 SME,而据我们所知,任何其他方法都无法获得 SME。我们使用密度泛函方法,在三种不同的理论水平上对 LPED 的计算进行了评估。在所研究的理论水平中,参考理论水平(ωB97X-D/aug-cc-pVTZ)和其他理论水平之间的 LPED 线性和 SME 相似。此外,LPED 被间接用于获得具有多重相互作用的复合物(如 EDTA-Ca+2 和磷霉素-Ca+2)分子间相互作用的局部结合能。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


