Computational analysis of natural compounds as potential phosphodiesterase type 5A inhibitors
IF 3.1
4区 生物学
Q2 BIOLOGY
引用次数: 0
Abstract
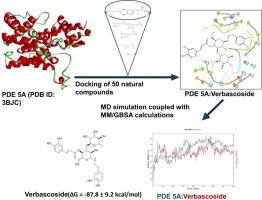
Phosphodiesterase type 5 (PDE5) is a cyclic nucleotide-hydrolyzing enzyme that plays essential roles in the regulation of second messenger cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) produced in response to various stimuli. Pharmacological inhibition of PDE5 has been shown to have several therapeutic uses, including treating cardiovascular diseases and erectile dysfunction. In search of PDE5A inhibitors with safer pharmacokinetic properties, computational analyses of the binding propensity of fifty natural compounds comprising flavonoids, polyphenols, and glycosides were conducted. Molecular dynamics simulation coupled with Molecular mechanics with generalized Born and surface area solvation (MM/GBSA) showed that verbascoside may inhibit the activity of PDE5 with a comparative binding energy (ΔG) of -87.8 ± 9.2 kcal/mol to that of the cocrystal ligand (PDB ID: 3BJC), having ΔG = -77.7±4.5 kcal/mol. However, the other top compounds studied were found to have lower binding propensities than the cocrystal ligand WAN: hesperidin (ΔG = -33.8 ± 3.4 kcal/mol), rutin (ΔG = -23.6 ± 26.3 kcal/mol), caftaric acid (ΔG = -21.2 ±3.6 kcal/mol), and chlorogenic acid (ΔG = 6.0 ± 16.5 kcal/mol). Therefore, verbascoside may serve as a potential PDE5A inhibitor while hesperidin, rutin, and caftaric acid may provide templates for further structural optimization for the designs of safer PDE5 inhibitors.
作为潜在 5A 型磷酸二酯酶抑制剂的天然化合物的计算分析
5 型磷酸二酯酶(PDE5)是一种环核苷酸水解酶,在调节第二信使环磷酸腺苷(cAMP)和环磷酸鸟苷(cGMP)对各种刺激的反应中发挥着重要作用。药理抑制 PDE5 已被证明具有多种治疗用途,包括治疗心血管疾病和勃起功能障碍。为了寻找具有更安全药代动力学特性的 PDE5A 抑制剂,研究人员对包括类黄酮、多酚和苷类在内的 50 种天然化合物的结合倾向进行了计算分析。分子动力学模拟结合广义玻恩和表面积溶解分子力学(MM/GBSA)表明,马鞭草苷可抑制 PDE5 的活性,其结合能(ΔG)为-87.8 ± 9.2 kcal/mol,而共晶体配体(PDB ID:3BJC)的结合能(ΔG = -77.7±4.5 kcal/mol)为-77.8 ± 9.2 kcal/mol。然而,研究发现其他顶级化合物的结合率低于共晶配体 WAN:橙皮甙(ΔG = -33.8 ± 3.4 kcal/mol)、芦丁(ΔG = -23.6 ± 26.3 kcal/mol)、茶醛酸(ΔG = -21.2 ± 3.6 kcal/mol)和绿原酸(ΔG = 6.0 ± 16.5 kcal/mol)。因此,马鞭草苷可作为一种潜在的 PDE5A 抑制剂,而橙皮甙、芦丁和茶黄酸则可为进一步优化结构以设计更安全的 PDE5 抑制剂提供模板。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational Biology and Chemistry
生物-计算机:跨学科应用
CiteScore
6.10
自引率
3.20%
发文量
142
审稿时长
24 days
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


