First-principles investigation of methane to methanol conversion on Ti2CO2 MXene supported single-atom catalyst
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Developing efficient catalysts for the conversion of methane (CH4) to methanol (CH3OH) remains a critical challenge in the chemical industry, with significant implications for both energy production and environmental sustainability. This study pioneers the exploration of the Sc/Ti-Ti2CO2 single-atom catalysts (SACs) for this transformation, utilizing density functional theory (DFT) calculations. Notably, our findings reveal that Sc and Ti are uniquely stable on the Ti2CO2 MXene surface, a discovery that could inform future catalyst designs. We also demonstrate that while CH4 weakly physisorbs on the Sc/Ti-Ti2CO2 surface, N2O molecules decompose directly into N2 and highly reactive O* species, which bind with Sc/Ti to drive the catalytic process. The oxidation of CH4 proceeds in two steps: CH4 + O* → CH3* + OH* with reaction barriers of 0.58 eV (Sc) and 1.38 eV (Ti), followed by CH3* + OH* → CH3OH with barriers of 1.5 eV (Sc) and 1.61 eV (Ti). Importantly, the low desorption energy of CH3OH, especially on Sc (0.85 eV), highlights the exceptional catalytic potential of Sc/Ti2CO2 for the direct conversion of CH4 to CH3OH. These results not only underscore the feasibility of using MXene-based SACs for CH4 oxidation but also provide a theoretical foundation for the development of highly efficient catalysts in this domain.
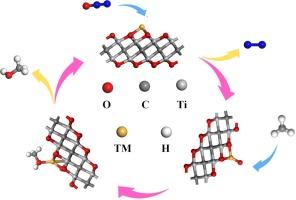
Ti2CO2 MXene 单原子支撑催化剂上甲烷到甲醇转化的第一性原理研究
开发将甲烷(CH4)转化为甲醇(CH3OH)的高效催化剂仍然是化学工业面临的一项重大挑战,对能源生产和环境可持续性都有重要影响。本研究利用密度泛函理论(DFT)计算,率先探索了用于这种转化的 Sc/Ti-Ti2CO2 单原子催化剂(SAC)。值得注意的是,我们的研究结果表明,Sc 和 Ti 在 Ti2CO2 MXene 表面具有独特的稳定性,这一发现可为未来的催化剂设计提供参考。我们还证明,虽然 CH4 在 Sc/Ti-Ti2CO2 表面上的物理吸附力很弱,但 N2O 分子会直接分解成 N2 和高活性的 O* 物种,它们与 Sc/Ti 结合,推动催化过程。CH4 的氧化过程分为两个步骤:CH4 + O* → CH3* + OH*,反应势垒分别为 0.58 eV(Sc)和 1.38 eV(Ti),然后是 CH3* + OH* → CH3OH,反应势垒分别为 1.5 eV(Sc)和 1.61 eV(Ti)。重要的是,CH3OH 的解吸能很低,尤其是在 Sc 上(0.85 eV),这凸显了 Sc/Ti2CO2 将 CH4 直接转化为 CH3OH 的巨大催化潜力。这些结果不仅强调了使用基于 MXene 的 SACs 进行 CH4 氧化的可行性,还为开发该领域的高效催化剂奠定了理论基础。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


