Zhi-Xin Bai, Fan-Jin Lu, Qi-Jun Liu, Zheng-Tang Liu
{"title":"Structural and Electronic Properties in Monolayer MoS \\({}_{\\mathbf{2}}\\) with Various Vacancies: First-Principles Calculations","authors":"Zhi-Xin Bai, Fan-Jin Lu, Qi-Jun Liu, Zheng-Tang Liu","doi":"10.3103/S0027134924700668","DOIUrl":null,"url":null,"abstract":"<p>The structural and electronic properties of perfect and defective MoS<span>\\({}_{2}\\)</span> have been calculated with first-principles density functional theory. The defect stability has been evaluated using the defect formation energy. The calculated results show that the formation of Mo, S, and Mo–S defects requires extra energy. Moreover, the S vacancy is energetically more favourable than the Mo vacancy. After doping, the defect level is introduced into the forbidden band. The defect level in the Mo defect model tends to be the acceptor level, while the defect level in the S defect model and Mo–S defect model tends to be the donor level. These three different defect models still maintain the direct band gap characteristics as perfect models. Then, the type of conduction after doping is also analyzed. The projected state density and the charge density of these four models have been analyzed, showing the positions of defect energy levels and change of chemical bonds.</p>","PeriodicalId":711,"journal":{"name":"Moscow University Physics Bulletin","volume":"79 4","pages":"500 - 506"},"PeriodicalIF":0.4000,"publicationDate":"2024-10-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Moscow University Physics Bulletin","FirstCategoryId":"101","ListUrlMain":"https://link.springer.com/article/10.3103/S0027134924700668","RegionNum":4,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"PHYSICS, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
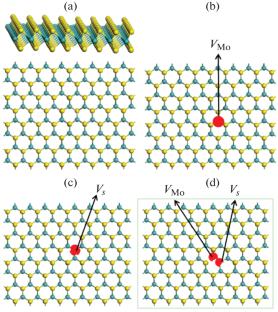
The structural and electronic properties of perfect and defective MoS\({}_{2}\) have been calculated with first-principles density functional theory. The defect stability has been evaluated using the defect formation energy. The calculated results show that the formation of Mo, S, and Mo–S defects requires extra energy. Moreover, the S vacancy is energetically more favourable than the Mo vacancy. After doping, the defect level is introduced into the forbidden band. The defect level in the Mo defect model tends to be the acceptor level, while the defect level in the S defect model and Mo–S defect model tends to be the donor level. These three different defect models still maintain the direct band gap characteristics as perfect models. Then, the type of conduction after doping is also analyzed. The projected state density and the charge density of these four models have been analyzed, showing the positions of defect energy levels and change of chemical bonds.
期刊介绍:
Moscow University Physics Bulletin publishes original papers (reviews, articles, and brief communications) in the following fields of experimental and theoretical physics: theoretical and mathematical physics; physics of nuclei and elementary particles; radiophysics, electronics, acoustics; optics and spectroscopy; laser physics; condensed matter physics; chemical physics, physical kinetics, and plasma physics; biophysics and medical physics; astronomy, astrophysics, and cosmology; physics of the Earth’s, atmosphere, and hydrosphere.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


