Huseyin Tunc, Sumeyye Yilmaz, Busra Nur Darendeli Kiraz, Murat Sari, Seyfullah Enes Kotil, Ozge Sensoy, Serdar Durdagi
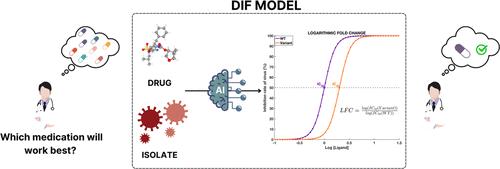
{"title":"Improving Predictive Efficacy for Drug Resistance in Novel HIV-1 Protease Inhibitors through Transfer Learning Mechanisms.","authors":"Huseyin Tunc, Sumeyye Yilmaz, Busra Nur Darendeli Kiraz, Murat Sari, Seyfullah Enes Kotil, Ozge Sensoy, Serdar Durdagi","doi":"10.1021/acs.jcim.4c01037","DOIUrl":null,"url":null,"abstract":"<p><p>The human immunodeficiency virus presents a significant global health challenge due to its rapid mutation and the development of resistance mechanisms against antiretroviral drugs. Recent studies demonstrate the impressive performance of machine learning (ML) and deep learning (DL) models in predicting the drug resistance profile of specific FDA-approved inhibitors. However, generalizing ML and DL models to learn not only from isolates but also from inhibitor representations remains challenging for HIV-1 infection. We propose a novel drug-isolate-fold change (DIF) model framework that aims to predict drug resistance score directly from the protein sequence and inhibitor representation. Various ML and DL models, inhibitor representations, and protein representations were analyzed through realistic validation mechanisms. To enhance the molecular learning capacity of DIF models, we employ a transfer learning approach by pretraining a graph neural network (GNN) model for activity prediction on a data set of 4855 HIV-1 protease inhibitors (PIs). By performing various realistic validation strategies on internal and external genotype-phenotype data sets, we statistically show that the learned representations of inhibitors improve the predictive ability of DIF-based ML and DL models. We achieved an accuracy of 0.802, AUROC of 0.874, and <i>r</i> of 0.727 for the unseen external PIs. By comparing the DIF-based models with a null model consisting of isolate-fold change (IF) architecture, it is observed that the DIF models significantly benefit from molecular representations. Combined results from various testing strategies and statistical tests confirm the effectiveness of DIF models in testing novel PIs for drug resistance in the presence of an isolate.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"7844-7863"},"PeriodicalIF":5.6000,"publicationDate":"2024-10-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c01037","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/11 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
The human immunodeficiency virus presents a significant global health challenge due to its rapid mutation and the development of resistance mechanisms against antiretroviral drugs. Recent studies demonstrate the impressive performance of machine learning (ML) and deep learning (DL) models in predicting the drug resistance profile of specific FDA-approved inhibitors. However, generalizing ML and DL models to learn not only from isolates but also from inhibitor representations remains challenging for HIV-1 infection. We propose a novel drug-isolate-fold change (DIF) model framework that aims to predict drug resistance score directly from the protein sequence and inhibitor representation. Various ML and DL models, inhibitor representations, and protein representations were analyzed through realistic validation mechanisms. To enhance the molecular learning capacity of DIF models, we employ a transfer learning approach by pretraining a graph neural network (GNN) model for activity prediction on a data set of 4855 HIV-1 protease inhibitors (PIs). By performing various realistic validation strategies on internal and external genotype-phenotype data sets, we statistically show that the learned representations of inhibitors improve the predictive ability of DIF-based ML and DL models. We achieved an accuracy of 0.802, AUROC of 0.874, and r of 0.727 for the unseen external PIs. By comparing the DIF-based models with a null model consisting of isolate-fold change (IF) architecture, it is observed that the DIF models significantly benefit from molecular representations. Combined results from various testing strategies and statistical tests confirm the effectiveness of DIF models in testing novel PIs for drug resistance in the presence of an isolate.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


