Justin A. Kaskow, Eric T. Hahnert, Thomas K. Porter, Yali Lu*, Valentin Stanev*, Chendi Niu, Wei Xu, Methal Albarghouthi and Chunlei Wang,
{"title":"Predicting Peptide Ionization Efficiencies for Electrospray Ionization Mass Spectrometry Using Machine Learning","authors":"Justin A. Kaskow, Eric T. Hahnert, Thomas K. Porter, Yali Lu*, Valentin Stanev*, Chendi Niu, Wei Xu, Methal Albarghouthi and Chunlei Wang, ","doi":"10.1021/jasms.4c0013710.1021/jasms.4c00137","DOIUrl":null,"url":null,"abstract":"<p >Mass spectrometry (MS) is inherently an information-rich technique. In this era of big data, label-free MS quantification for nontargeted studies has gained increasing popularity, especially for complex systems. One of the cornerstones of successful label-free quantification is the predictive modeling of ionization efficiency (IE) based on solutes’ physicochemical properties. While many have studied IE modeling for small molecules, there are limited reports on peptide IEs. In this study, we leverage the stoichiometric relationship in trypsin digests of well-characterized monoclonal antibodies (mAbs) to compile a data set of relative ionization efficiencies (RIEs) for 241 peptides. From each peptide’s sequence, we computed a set of physiochemical descriptors, which were then used to train machine learning regression models to predict RIEs. Peptides shorter than 20 amino acids had RIEs that were highly correlated to their molecular weight. A random forest (RF) model was able to best predict the RIEs of a test data set with a mean relative error of 23.9%. For larger peptides, a multilayer perceptron (MLP) model improved RIE prediction compared to current best practices, reducing mean relative error from 60.5% to 32.0%. Finally, we also show the application of the RF model in label-free relative protein quantification and improving the quantification of peptide post-translational modifications (PTMs). This approach to predicting peptide IEs from their sequences enables the development of accurate label-free quantification workflows for peptide and protein analysis.</p>","PeriodicalId":672,"journal":{"name":"Journal of the American Society for Mass Spectrometry","volume":null,"pages":null},"PeriodicalIF":3.1000,"publicationDate":"2024-09-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Society for Mass Spectrometry","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/jasms.4c00137","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
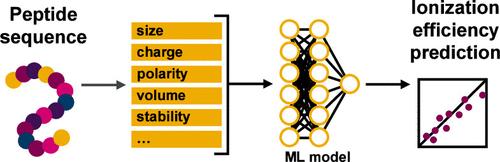
Mass spectrometry (MS) is inherently an information-rich technique. In this era of big data, label-free MS quantification for nontargeted studies has gained increasing popularity, especially for complex systems. One of the cornerstones of successful label-free quantification is the predictive modeling of ionization efficiency (IE) based on solutes’ physicochemical properties. While many have studied IE modeling for small molecules, there are limited reports on peptide IEs. In this study, we leverage the stoichiometric relationship in trypsin digests of well-characterized monoclonal antibodies (mAbs) to compile a data set of relative ionization efficiencies (RIEs) for 241 peptides. From each peptide’s sequence, we computed a set of physiochemical descriptors, which were then used to train machine learning regression models to predict RIEs. Peptides shorter than 20 amino acids had RIEs that were highly correlated to their molecular weight. A random forest (RF) model was able to best predict the RIEs of a test data set with a mean relative error of 23.9%. For larger peptides, a multilayer perceptron (MLP) model improved RIE prediction compared to current best practices, reducing mean relative error from 60.5% to 32.0%. Finally, we also show the application of the RF model in label-free relative protein quantification and improving the quantification of peptide post-translational modifications (PTMs). This approach to predicting peptide IEs from their sequences enables the development of accurate label-free quantification workflows for peptide and protein analysis.
期刊介绍:
The Journal of the American Society for Mass Spectrometry presents research papers covering all aspects of mass spectrometry, incorporating coverage of fields of scientific inquiry in which mass spectrometry can play a role.
Comprehensive in scope, the journal publishes papers on both fundamentals and applications of mass spectrometry. Fundamental subjects include instrumentation principles, design, and demonstration, structures and chemical properties of gas-phase ions, studies of thermodynamic properties, ion spectroscopy, chemical kinetics, mechanisms of ionization, theories of ion fragmentation, cluster ions, and potential energy surfaces. In addition to full papers, the journal offers Communications, Application Notes, and Accounts and Perspectives

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


