Elisa Lamback, Renan Lyra Miranda, Leila Chimelli, Felipe Andreiuolo, Leandro Kasuki, Luiz Eduardo Wildemberg, Mônica R Gadelha
{"title":"Pituitary gigantism due to a novel <i>AIP</i> germline splice-site variant.","authors":"Elisa Lamback, Renan Lyra Miranda, Leila Chimelli, Felipe Andreiuolo, Leandro Kasuki, Luiz Eduardo Wildemberg, Mônica R Gadelha","doi":"10.1530/EO-24-0003","DOIUrl":null,"url":null,"abstract":"<p><p>Pituitary gigantism is a rare pediatric disorder caused by excess growth hormone (GH) secretion. In almost 50% of cases, a genetic cause can be identified, with pathogenic variants in the aryl hydrocarbon receptor-interacting protein (<i>AIP</i>) gene being the most common. We present a case of an 11-year-old boy who exhibited progressive vision loss, associated with accelerated linear growth, and weight gain. On physical examination, he had enlarged hands, right eye amaurosis, and was already above his target height. Increased GH and IGF-I concentrations confirmed the diagnosis of pituitary gigantism. Magnetic resonance imaging showed a giant sellar lesion with supra- and para-sellar extensions. He underwent two surgeries which did not achieve a cure or visual improvement. Histopathological analysis revealed a sparsely granulated tumor, negative for somatostatin receptor type 2 (SST2) and an immunoreactivity score of 6 for somatostatin receptor type 5 (SST5). Our published artificial intelligence prediction model predicted an 83% chance of not responding to first-generation somatostatin receptor ligands. Pasireotide was therefore prescribed, and afterward cabergoline was added on. IGF-I concentrations decreased but did not normalize. We discovered a novel germline single nucleotide variant in the splicing donor region of intron 2 of the <i>AIP</i> gene (NM_003977.4:c.279+1 G>A), classified as likely pathogenic according to the American College of Medical Genetics and Genomics guidelines.</p>","PeriodicalId":72907,"journal":{"name":"Endocrine oncology (Bristol, England)","volume":"4 1","pages":"e240003"},"PeriodicalIF":0.0000,"publicationDate":"2024-09-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11466259/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Endocrine oncology (Bristol, England)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1530/EO-24-0003","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
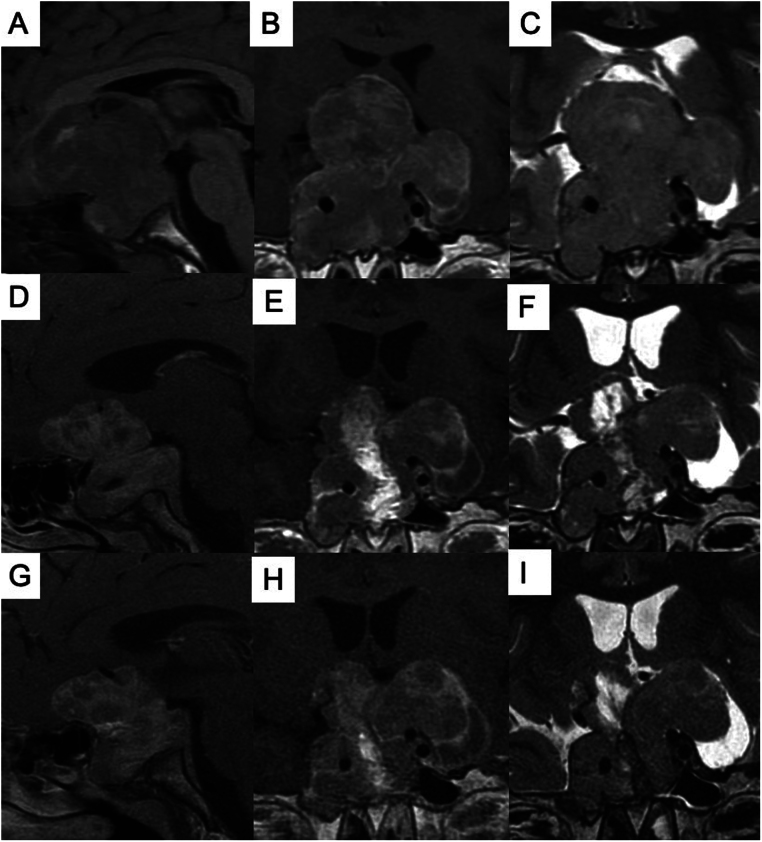
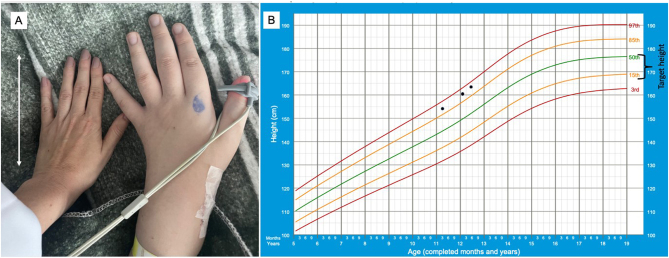
Pituitary gigantism is a rare pediatric disorder caused by excess growth hormone (GH) secretion. In almost 50% of cases, a genetic cause can be identified, with pathogenic variants in the aryl hydrocarbon receptor-interacting protein (AIP) gene being the most common. We present a case of an 11-year-old boy who exhibited progressive vision loss, associated with accelerated linear growth, and weight gain. On physical examination, he had enlarged hands, right eye amaurosis, and was already above his target height. Increased GH and IGF-I concentrations confirmed the diagnosis of pituitary gigantism. Magnetic resonance imaging showed a giant sellar lesion with supra- and para-sellar extensions. He underwent two surgeries which did not achieve a cure or visual improvement. Histopathological analysis revealed a sparsely granulated tumor, negative for somatostatin receptor type 2 (SST2) and an immunoreactivity score of 6 for somatostatin receptor type 5 (SST5). Our published artificial intelligence prediction model predicted an 83% chance of not responding to first-generation somatostatin receptor ligands. Pasireotide was therefore prescribed, and afterward cabergoline was added on. IGF-I concentrations decreased but did not normalize. We discovered a novel germline single nucleotide variant in the splicing donor region of intron 2 of the AIP gene (NM_003977.4:c.279+1 G>A), classified as likely pathogenic according to the American College of Medical Genetics and Genomics guidelines.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


