Maximilian Bschorer, Matthias Dottermusch, Jakob Matschke, Jens Gempt, Ulrich Schüller, Malte Mohme
{"title":"40-Year-old man with two asynchronous spinal cord tumors","authors":"Maximilian Bschorer, Matthias Dottermusch, Jakob Matschke, Jens Gempt, Ulrich Schüller, Malte Mohme","doi":"10.1111/bpa.13309","DOIUrl":null,"url":null,"abstract":"<p>A 38-year-old individual presented with intermittent bladder dysfunction, radicular pain, and mild foot elevator paresis. After an emergency lumbar MRI revealed a vertebral disc herniation at the L5/S1 level, an emergency sequestrectomy was performed. Although the radicular pain resolved postoperatively, the patient's bladder voiding dysfunction worsened. Further MRI imaging revealed a contrast-enhancing intramedullary lesion at the TH11/TH12 spinal levels (Figure 1A).</p><p>The patient was transferred to the neurosurgical department for further treatment, and urgent surgery was performed with continuous intraoperative neurophysiological monitoring. Histopathological analysis was conducted and subsequent DNA methylation analysis confirmed the histological diagnosis. The patient did not undergo genetic testing for neurofibromatosis (NF), as he did not exhibit any other typical features or tumors associated with NF.</p><p>Postoperative paraparesis and bladder dysfunction were promptly resolved in neurological rehabilitation, resulting in a McCormick score of one. Three years later, the patient presented with new radiculopathy in both legs accompanied and hypesthesia. MRI showed a new contrast-enhancing lesion at L2/L3, which was located at a distance from the previous lesion (Figure 1B). The patient underwent surgery for this symptomatic lesion. The histopathological and methylation analysis revealed a distinct diagnosis, when compared to the histological examination of the initial tumor. Postoperative imaging showed no residual tumor, and there were no other central nervous system (CNS) manifestations of the tumor. The patient's rapid recovery from neurological symptoms allowed for discharge to ambulatory service.</p><p>Histological comparison of the first and second tumor revealed distinctive features. The first tumor was characterized by isomorphic glial cells with round nuclei within a delicate fibrillary matrix, as observed in H&E staining (Figure 2, Box 1). The tumor cells showed clear expression of glial fibrillary acidic protein (GFAP), but not OLIG2 or NMYC. The Ki67 labeling index was below 5% of the tumor cell nuclei. Zones without nuclei around blood vessels, known as perivascular pseudorosettes, were also present.</p><p>The second tumor was characterized by more spindled tumor cells within a coarse myxoid glial matrix, indicative of a divergent histological diagnosis. The cell nuclei were oval to elongated with slightly loosened nuclear chromatin. Similar to the first tumor, strong GFAP immunostaining was observed. The second tumor was distinctively marked by nuclear expression of HOXB1, absent in the first tumor.</p><p>For both samples, DNA methylation data were obtained using the Illumina Human MethylationEPIC (850 k) array bead chips. The classification of brain tumors based on DNA methylation was performed using the publicly available “classifier” tool, version v12.8 (www.molecularneuropathology.org/mnp). The epigenetic analysis confirmed both histological diagnoses with a high concordance, reflected by a matching score of 0.99.</p><p>Concurrent diagnosis of spinal ependymoma (WHO-CNS Grade 2) and myxopapillary ependymoma (WHO-CNS Grade 2).</p><p>Rare primary tumors of the CNS, such as spinal ependymomas (WHO-CNS Grade 2) (SPE) and myxopapillary ependymomas (WHO-CNS Grade 2) (MPE), are classified by the 2021 World Health Organization (WHO) classification of CNS tumors. The classification of CNS tumors is constantly under review and undergoes periodic updates to incorporate novel molecular and pathological data [<span>1</span>]. MPE and SPE have historically been challenging to differentiate histologically and immunohistochemically, emphasizing the importance of molecular analysis of these tumors [<span>2</span>]. Witt et al. highlighted this in their 2018 study, which demonstrated a high rate of reclassification among spinal ependymomas based on their molecular characteristics [<span>2</span>]. Recent advancements have enabled the use of HOXB13 gene expression staining as a reliable method for immunohistochemical differentiation among these tumors [<span>3</span>].</p><p>Individuals with neurofibromatosis type 2 (NF2) are genetically predisposed to develop ependymomas of various histology and location. On the other hand, MPEs are often diagnosed in younger adults and are typically located in the cauda equina and medullary conus. While MPE are known to potentially cause local relapse and even distant spinal seeding in rare cases (PMID: 2187161), metastatic seeding of SPE outside the primary intramedullary location is extremely rare (PMID: 37260799).</p><p>Initially classifying this patient as having metastatic disease might have led to recommending radiosurgery. However, this case emphasizes the necessity of surgical intervention over radiosurgery. Although gross total resection is the ideal treatment for these tumor types, the proximity to critical neurological structures often limits this to subtotal resection or biopsy. Thus, radiotherapy is frequently recommended when surgical options are limited, but its efficacy remains under continuous evaluation [<span>2</span>]. Given the heterogeneous nature of spinal ependymomas' histological appearance, this case reinforces the reliance on molecular diagnostics to guide therapeutic decisions.</p><p>MB drafted the manuscript. MD prepared the slides and performed the histological and immunohistological analysis. MM and US revised the manuscript. MM, US, JM and JG contributed to the study design and supported the process of drafting and submitting the manuscript.</p><p>U. S. and M. M. are supported by the DFG (German Research Foundation). U. S. is also supported by the Fördergemeinschaft Kinderkrebszentrum Hamburg.</p><p>The authors declare no conflicts of interest.</p><p>The patient consented to be included in this publication.</p>","PeriodicalId":9290,"journal":{"name":"Brain Pathology","volume":"34 6","pages":""},"PeriodicalIF":6.2000,"publicationDate":"2024-10-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/bpa.13309","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain Pathology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/bpa.13309","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
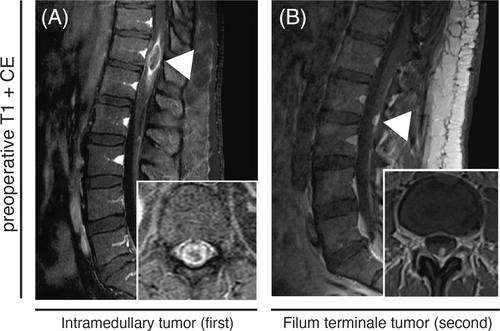
A 38-year-old individual presented with intermittent bladder dysfunction, radicular pain, and mild foot elevator paresis. After an emergency lumbar MRI revealed a vertebral disc herniation at the L5/S1 level, an emergency sequestrectomy was performed. Although the radicular pain resolved postoperatively, the patient's bladder voiding dysfunction worsened. Further MRI imaging revealed a contrast-enhancing intramedullary lesion at the TH11/TH12 spinal levels (Figure 1A).
The patient was transferred to the neurosurgical department for further treatment, and urgent surgery was performed with continuous intraoperative neurophysiological monitoring. Histopathological analysis was conducted and subsequent DNA methylation analysis confirmed the histological diagnosis. The patient did not undergo genetic testing for neurofibromatosis (NF), as he did not exhibit any other typical features or tumors associated with NF.
Postoperative paraparesis and bladder dysfunction were promptly resolved in neurological rehabilitation, resulting in a McCormick score of one. Three years later, the patient presented with new radiculopathy in both legs accompanied and hypesthesia. MRI showed a new contrast-enhancing lesion at L2/L3, which was located at a distance from the previous lesion (Figure 1B). The patient underwent surgery for this symptomatic lesion. The histopathological and methylation analysis revealed a distinct diagnosis, when compared to the histological examination of the initial tumor. Postoperative imaging showed no residual tumor, and there were no other central nervous system (CNS) manifestations of the tumor. The patient's rapid recovery from neurological symptoms allowed for discharge to ambulatory service.
Histological comparison of the first and second tumor revealed distinctive features. The first tumor was characterized by isomorphic glial cells with round nuclei within a delicate fibrillary matrix, as observed in H&E staining (Figure 2, Box 1). The tumor cells showed clear expression of glial fibrillary acidic protein (GFAP), but not OLIG2 or NMYC. The Ki67 labeling index was below 5% of the tumor cell nuclei. Zones without nuclei around blood vessels, known as perivascular pseudorosettes, were also present.
The second tumor was characterized by more spindled tumor cells within a coarse myxoid glial matrix, indicative of a divergent histological diagnosis. The cell nuclei were oval to elongated with slightly loosened nuclear chromatin. Similar to the first tumor, strong GFAP immunostaining was observed. The second tumor was distinctively marked by nuclear expression of HOXB1, absent in the first tumor.
For both samples, DNA methylation data were obtained using the Illumina Human MethylationEPIC (850 k) array bead chips. The classification of brain tumors based on DNA methylation was performed using the publicly available “classifier” tool, version v12.8 (www.molecularneuropathology.org/mnp). The epigenetic analysis confirmed both histological diagnoses with a high concordance, reflected by a matching score of 0.99.
Concurrent diagnosis of spinal ependymoma (WHO-CNS Grade 2) and myxopapillary ependymoma (WHO-CNS Grade 2).
Rare primary tumors of the CNS, such as spinal ependymomas (WHO-CNS Grade 2) (SPE) and myxopapillary ependymomas (WHO-CNS Grade 2) (MPE), are classified by the 2021 World Health Organization (WHO) classification of CNS tumors. The classification of CNS tumors is constantly under review and undergoes periodic updates to incorporate novel molecular and pathological data [1]. MPE and SPE have historically been challenging to differentiate histologically and immunohistochemically, emphasizing the importance of molecular analysis of these tumors [2]. Witt et al. highlighted this in their 2018 study, which demonstrated a high rate of reclassification among spinal ependymomas based on their molecular characteristics [2]. Recent advancements have enabled the use of HOXB13 gene expression staining as a reliable method for immunohistochemical differentiation among these tumors [3].
Individuals with neurofibromatosis type 2 (NF2) are genetically predisposed to develop ependymomas of various histology and location. On the other hand, MPEs are often diagnosed in younger adults and are typically located in the cauda equina and medullary conus. While MPE are known to potentially cause local relapse and even distant spinal seeding in rare cases (PMID: 2187161), metastatic seeding of SPE outside the primary intramedullary location is extremely rare (PMID: 37260799).
Initially classifying this patient as having metastatic disease might have led to recommending radiosurgery. However, this case emphasizes the necessity of surgical intervention over radiosurgery. Although gross total resection is the ideal treatment for these tumor types, the proximity to critical neurological structures often limits this to subtotal resection or biopsy. Thus, radiotherapy is frequently recommended when surgical options are limited, but its efficacy remains under continuous evaluation [2]. Given the heterogeneous nature of spinal ependymomas' histological appearance, this case reinforces the reliance on molecular diagnostics to guide therapeutic decisions.
MB drafted the manuscript. MD prepared the slides and performed the histological and immunohistological analysis. MM and US revised the manuscript. MM, US, JM and JG contributed to the study design and supported the process of drafting and submitting the manuscript.
U. S. and M. M. are supported by the DFG (German Research Foundation). U. S. is also supported by the Fördergemeinschaft Kinderkrebszentrum Hamburg.
The authors declare no conflicts of interest.
The patient consented to be included in this publication.
期刊介绍:
Brain Pathology is the journal of choice for biomedical scientists investigating diseases of the nervous system. The official journal of the International Society of Neuropathology, Brain Pathology is a peer-reviewed quarterly publication that includes original research, review articles and symposia focuses on the pathogenesis of neurological disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


