Minji Lee, Myung Jin Son, Sin-Hyoung Hong, Jae-Sung Ryu, Ji-Hyeon Min, Dong-Eon Lee, Ji Hoon Lee, Nam Doo Kim, Shi-Young Park, Darong Kim, Jeongmin Joo, Jisung Kwak, Kook Hwan Kim, Yong-Ho Lee, Byeong-Rak Keum, Hyun Seok Song, Youngae Jung, Koon Soon Kim, Gun-Hwa Kim
{"title":"Discovery of a selective cytochrome P450 4A inhibitor for the treatment of metabolic dysfunction-associated fatty liver disease","authors":"Minji Lee, Myung Jin Son, Sin-Hyoung Hong, Jae-Sung Ryu, Ji-Hyeon Min, Dong-Eon Lee, Ji Hoon Lee, Nam Doo Kim, Shi-Young Park, Darong Kim, Jeongmin Joo, Jisung Kwak, Kook Hwan Kim, Yong-Ho Lee, Byeong-Rak Keum, Hyun Seok Song, Youngae Jung, Koon Soon Kim, Gun-Hwa Kim","doi":"10.1002/ctm2.1816","DOIUrl":null,"url":null,"abstract":"<p>Dear Editor,</p><p>Metabolic dysfunction-associated fatty liver disease (MAFLD), a revised definition of nonalcoholic fatty liver disease (NAFLD), comprises patients with hepatic steatosis who fulfil the criteria of overweight/obesity, type II diabetes mellitus (T2DM), or more than two metabolic abnormalities,<span><sup>1</sup></span> providing a valuable tool for identifying patients with fatty liver at higher risk of disease progression.<span><sup>2</sup></span> MAFLD is a complex disease in which various pathogenic factors contribute to its progression, including fat accumulation, lipotoxicity, oxidative stress, and endoplasmic reticulum (ER) stress. The heterogeneous risk profile of MAFLD presents challenges for effective treatment. Within the mammalian liver, cytochrome P450 4A (CYP4A) functions as a fatty acid hydroxylase, actively participating in oxidative metabolism and catalyzing the breakdown of lipid peroxides, consequently generating reactive oxygen species (ROS). Previous research has demonstrated that targeting CYP4A shows potential in exploring the pathophysiology of liver diseases, including MAFLD and diabetes.<span><sup>3-5</sup></span> Taking these findings together, we suggest that CYP4A holds significant promise for the treatment of MAFLD, and the discovery of a CYP4A inhibitor may serve as a potent drug candidate.</p><p>We identified CYP4A inhibitors through in silico analysis. Among several hit compounds, C418 significantly reduced CYP4A enzyme activity. Subsequently, we synthesized derivatives, namely C4181 (C1) and C4182 (C2) (Figure 1A). Both C1 and C2 exhibited substantial reduction at a dose of 5 µM (Figures S1A and S2A), demonstrating potent inhibition with IC<sub>50</sub> (Figure 1B). Importantly, neither compound exhibited cytotoxic effects (Figure S1B,C) and demonstrated drug-like properties in various assays, including CYP inhibition, metabolic stability, plasma stability, and permeability (Tables S1 and S2).</p><p>We investigated the effects of novel CYP4A inhibitors using HepG2 cells exposed to lipid overload or induced ER stress. C1 and C2 increased glucose uptake (Figures S1D and S2B) and significantly mitigated lipid accumulation and ROS generation (Figures S1E,F and S2C,D). Furthermore, they restored the expression of proteins implicated in gluconeogenesis, lipogenesis, and ER stress (Figures S1G-K and S2E), indicating the effectiveness of CYP4A inhibitors in in vitro models. Additionally, we determine the effect of C1 and C2 in various in vivo models, encompassing diet-induced T2DM, <i>db/db</i> mice, and metabolic dysfunction-associated steatohepatitis (MASH). In the diet-induced T2DM model, we determined the optimal concentration of C1 and C2 (Figure S3) and observed a significant reduction in body mass without affecting food intake (Figure 1C,D). Glucose tolerance and insulin tolerance testing (ITT) exhibited significant improvements (Figure 1E), and serum glucose and insulin concentrations were abrogated (Table S3). Histological analysis revealed improved hepatic steatosis, with significant reductions in both hepatic and serum triglyceride (TG) concentrations and CD36 expression (Figure 1F–H, Figure S4A and Table S3). Moreover, C1 and C2 treatment decreased the concentration of hepatic malondialdehyde (Figure 1I) and 20-HETE, which is generated as a product of CYP4A enzymatic action (Figure 1J).<span><sup>6</sup></span> The treatments also improved serum concentrations of aspartate aminotransferase (AST), alanine transaminase (ALT), total cholesterol, LDL-cholesterol, LDL/HDL-cholesterol ratio and adiponectin (Table S3). Notably, the expression of mediators of gluconeogenesis, lipogenesis, ER stress, apoptosis, and insulin resistance was significantly restored (Figure 1K,L and Figure S4B,C). We next assessed the effects of C1 and C2 administration in <i>db/db</i> mice, which showed improvements in metabolic defects, ER stress, apoptosis, and insulin resistance, consistent with the effects observed in the diet-induced T2DM model (Figure S5).</p><p>Finally, we assessed the effect of C1 and C2 in the MASH model, induced by a methionine-choline-deficient (MCD) diet using <i>ob/ob</i> mice to replicate MASH histology (Figure 2A).<span><sup>7</sup></span> Although the MCD diet resulted in reductions in body weight over time (Figure S6), the CYP4A inhibitors showed significant improvement effects on MASH. They significantly improved serum glucose concentrations (Figure 2B), as well as both their serum and hepatic TG concentrations (Figure 2C,D). Histological examination demonstrated a significant reduction in intracytoplasmic lipid accumulation and collagen deposition (Figure 2E). Additionally, there was a decrease in hepatic 20-HETE concentration (Figure 2F), and serum AST and ALT activities were nearly normalized (Figure 2G), suggesting that C1 and C2 alleviate hepatic steatosis and liver damage. Furthermore, the expression of genes related to hepatic inflammation and fibrosis was significantly lower in the treated mice (Figure 2H,I). Therefore, C1 and C2 ameliorate steatosis and MASH.</p><p>Recently, it has been reported that patients with MAFLD,<span><sup>5, 8</sup></span> exhibit elevated expression of CYP4A in the liver, as was also observed in Korean patients (Figure S7). However, investigating the direct effect of CYP4A inhibitors in humans is challenging. To overcome this limitation, three-dimensional models were utilized as effective tools for preclinical drug discovery. We employed established models, including organoid-based and 3D HepaRG models of liver steatosis.<span><sup>9, 10</sup></span> Treatment with compounds C1 and C2 notably decreased the elevated mRNA expression and activity of CYP4A (Figure 3A,B and Figure S8A,B), improved glucose uptake and consumption (Figure 3C–E and Figure S8C), reduced lipid accumulation (Figure 3F–H and Figure S8D) and lowered triglyceride levels (Figure 3I and Figure S8E). Moreover, the expression of CD36 (Figure 3J) and various mediators related to lipogenesis, gluconeogenesis, ER stress and insulin resistance were downregulated by C1 and C2 in both organoid-based (Figure 3K–N and Figure S9) and 3D HepaRG models (Figure S8F,G).</p><p>To understand the mechanism of C1 and C2, we conducted transcriptomic analysis, focusing on differentially expressed genes and pathway analysis (Figure S10A–D and Table S4). Utilizing Ingenuity Pathway Analysis and Gene Set Enrichment Analysis, we observed a significant reduction in the expression of genes associated with hepatic steatosis, inflammation, T2DM, and MAFLD upon treatment with C1 and C2 (Figure 4A–D). Transcriptomic profiling further indicated a decrease in inflammation and ER stress-related genes following treatment with the CYP4A inhibitors (Figure 4E,F and Figure S10E,F). C1 and C2 also present similar downregulated genes related to MAFLD (Table S5). Collectively, these data demonstrate that inhibiting CYP4A effectively improves MAFLD by reducing hepatic steatosis, inflammation, and fibrosis, while enhancing ITT.</p><p>In conclusion, our findings highlight the promising therapeutic potential of CYP4A inhibitors in addressing MAFLD. These inhibitors operate through three distinct mechanisms: ER stress/oxidative stress, Fatty acid translocase (FAT/CD36)/lipotoxicity, and inflammation/fibrosis (Figure 4G). Our results demonstrate that these inhibitors could serve as innovative candidates, introducing a novel concept in the medical field for the treatment of MAFLD.</p><p>Minji Lee, Myung Jin Son and Sin-Hyoung Hong contributed equally to this work. Conceptualization: Minji Lee, Myung Jin Son and Gun-Hwa Kim; Methodology: Sin-Hyoung Hong, Jisung Kwak, Kook Hwan Kim, Yong-Ho Lee and Hyun Seok Song; Investigation: Minji Lee, Myung Jin Son, Sin-Hyoung Hong, Jae-Sung Ryu, Ji-Hyeon Min and Dong-Eon Lee; Visualization: Minji Lee, Myung Jin Son and Byeong-Rak Keum; Formal analysis: Minji Lee, Myung Jin Son, Ji Hoon Lee, Nam Doo Kim, Shi-Young Park, Darong Kim and Jeongmin Joo, Youngae Jung; Resources: Ji Hoon Lee, Nam Doo Kim, Darong Kim and Jeongmin Joo; Funding acquisition: Gun-Hwa Kim; Project administration: Koon Soon Kim and Gun-Hwa Kim; Supervision: Gun-Hwa Kim; Writing—original draft: Minji Lee; Writing—review & editing: Gun-Hwa Kim. All authors have read and approved the article.</p><p>The authors declare no conflict of interest.</p><p>This work was supported by grants from the Korea Basic Science Institute (Grant number C270300) and the National Research Foundation of Korea (Grant number NRF-2021R1A2C1008663).</p>","PeriodicalId":10189,"journal":{"name":"Clinical and Translational Medicine","volume":"14 10","pages":""},"PeriodicalIF":6.8000,"publicationDate":"2024-10-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11452733/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical and Translational Medicine","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ctm2.1816","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
Dear Editor,
Metabolic dysfunction-associated fatty liver disease (MAFLD), a revised definition of nonalcoholic fatty liver disease (NAFLD), comprises patients with hepatic steatosis who fulfil the criteria of overweight/obesity, type II diabetes mellitus (T2DM), or more than two metabolic abnormalities,1 providing a valuable tool for identifying patients with fatty liver at higher risk of disease progression.2 MAFLD is a complex disease in which various pathogenic factors contribute to its progression, including fat accumulation, lipotoxicity, oxidative stress, and endoplasmic reticulum (ER) stress. The heterogeneous risk profile of MAFLD presents challenges for effective treatment. Within the mammalian liver, cytochrome P450 4A (CYP4A) functions as a fatty acid hydroxylase, actively participating in oxidative metabolism and catalyzing the breakdown of lipid peroxides, consequently generating reactive oxygen species (ROS). Previous research has demonstrated that targeting CYP4A shows potential in exploring the pathophysiology of liver diseases, including MAFLD and diabetes.3-5 Taking these findings together, we suggest that CYP4A holds significant promise for the treatment of MAFLD, and the discovery of a CYP4A inhibitor may serve as a potent drug candidate.
We identified CYP4A inhibitors through in silico analysis. Among several hit compounds, C418 significantly reduced CYP4A enzyme activity. Subsequently, we synthesized derivatives, namely C4181 (C1) and C4182 (C2) (Figure 1A). Both C1 and C2 exhibited substantial reduction at a dose of 5 µM (Figures S1A and S2A), demonstrating potent inhibition with IC50 (Figure 1B). Importantly, neither compound exhibited cytotoxic effects (Figure S1B,C) and demonstrated drug-like properties in various assays, including CYP inhibition, metabolic stability, plasma stability, and permeability (Tables S1 and S2).
We investigated the effects of novel CYP4A inhibitors using HepG2 cells exposed to lipid overload or induced ER stress. C1 and C2 increased glucose uptake (Figures S1D and S2B) and significantly mitigated lipid accumulation and ROS generation (Figures S1E,F and S2C,D). Furthermore, they restored the expression of proteins implicated in gluconeogenesis, lipogenesis, and ER stress (Figures S1G-K and S2E), indicating the effectiveness of CYP4A inhibitors in in vitro models. Additionally, we determine the effect of C1 and C2 in various in vivo models, encompassing diet-induced T2DM, db/db mice, and metabolic dysfunction-associated steatohepatitis (MASH). In the diet-induced T2DM model, we determined the optimal concentration of C1 and C2 (Figure S3) and observed a significant reduction in body mass without affecting food intake (Figure 1C,D). Glucose tolerance and insulin tolerance testing (ITT) exhibited significant improvements (Figure 1E), and serum glucose and insulin concentrations were abrogated (Table S3). Histological analysis revealed improved hepatic steatosis, with significant reductions in both hepatic and serum triglyceride (TG) concentrations and CD36 expression (Figure 1F–H, Figure S4A and Table S3). Moreover, C1 and C2 treatment decreased the concentration of hepatic malondialdehyde (Figure 1I) and 20-HETE, which is generated as a product of CYP4A enzymatic action (Figure 1J).6 The treatments also improved serum concentrations of aspartate aminotransferase (AST), alanine transaminase (ALT), total cholesterol, LDL-cholesterol, LDL/HDL-cholesterol ratio and adiponectin (Table S3). Notably, the expression of mediators of gluconeogenesis, lipogenesis, ER stress, apoptosis, and insulin resistance was significantly restored (Figure 1K,L and Figure S4B,C). We next assessed the effects of C1 and C2 administration in db/db mice, which showed improvements in metabolic defects, ER stress, apoptosis, and insulin resistance, consistent with the effects observed in the diet-induced T2DM model (Figure S5).
Finally, we assessed the effect of C1 and C2 in the MASH model, induced by a methionine-choline-deficient (MCD) diet using ob/ob mice to replicate MASH histology (Figure 2A).7 Although the MCD diet resulted in reductions in body weight over time (Figure S6), the CYP4A inhibitors showed significant improvement effects on MASH. They significantly improved serum glucose concentrations (Figure 2B), as well as both their serum and hepatic TG concentrations (Figure 2C,D). Histological examination demonstrated a significant reduction in intracytoplasmic lipid accumulation and collagen deposition (Figure 2E). Additionally, there was a decrease in hepatic 20-HETE concentration (Figure 2F), and serum AST and ALT activities were nearly normalized (Figure 2G), suggesting that C1 and C2 alleviate hepatic steatosis and liver damage. Furthermore, the expression of genes related to hepatic inflammation and fibrosis was significantly lower in the treated mice (Figure 2H,I). Therefore, C1 and C2 ameliorate steatosis and MASH.
Recently, it has been reported that patients with MAFLD,5, 8 exhibit elevated expression of CYP4A in the liver, as was also observed in Korean patients (Figure S7). However, investigating the direct effect of CYP4A inhibitors in humans is challenging. To overcome this limitation, three-dimensional models were utilized as effective tools for preclinical drug discovery. We employed established models, including organoid-based and 3D HepaRG models of liver steatosis.9, 10 Treatment with compounds C1 and C2 notably decreased the elevated mRNA expression and activity of CYP4A (Figure 3A,B and Figure S8A,B), improved glucose uptake and consumption (Figure 3C–E and Figure S8C), reduced lipid accumulation (Figure 3F–H and Figure S8D) and lowered triglyceride levels (Figure 3I and Figure S8E). Moreover, the expression of CD36 (Figure 3J) and various mediators related to lipogenesis, gluconeogenesis, ER stress and insulin resistance were downregulated by C1 and C2 in both organoid-based (Figure 3K–N and Figure S9) and 3D HepaRG models (Figure S8F,G).
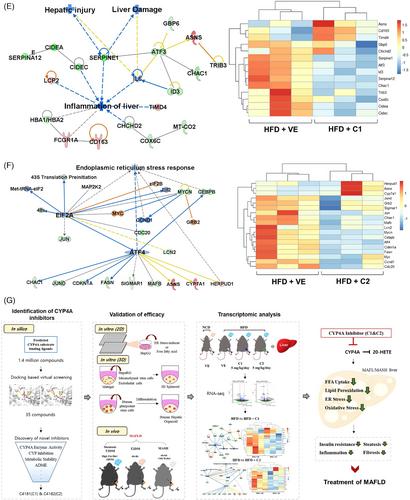
To understand the mechanism of C1 and C2, we conducted transcriptomic analysis, focusing on differentially expressed genes and pathway analysis (Figure S10A–D and Table S4). Utilizing Ingenuity Pathway Analysis and Gene Set Enrichment Analysis, we observed a significant reduction in the expression of genes associated with hepatic steatosis, inflammation, T2DM, and MAFLD upon treatment with C1 and C2 (Figure 4A–D). Transcriptomic profiling further indicated a decrease in inflammation and ER stress-related genes following treatment with the CYP4A inhibitors (Figure 4E,F and Figure S10E,F). C1 and C2 also present similar downregulated genes related to MAFLD (Table S5). Collectively, these data demonstrate that inhibiting CYP4A effectively improves MAFLD by reducing hepatic steatosis, inflammation, and fibrosis, while enhancing ITT.
In conclusion, our findings highlight the promising therapeutic potential of CYP4A inhibitors in addressing MAFLD. These inhibitors operate through three distinct mechanisms: ER stress/oxidative stress, Fatty acid translocase (FAT/CD36)/lipotoxicity, and inflammation/fibrosis (Figure 4G). Our results demonstrate that these inhibitors could serve as innovative candidates, introducing a novel concept in the medical field for the treatment of MAFLD.
Minji Lee, Myung Jin Son and Sin-Hyoung Hong contributed equally to this work. Conceptualization: Minji Lee, Myung Jin Son and Gun-Hwa Kim; Methodology: Sin-Hyoung Hong, Jisung Kwak, Kook Hwan Kim, Yong-Ho Lee and Hyun Seok Song; Investigation: Minji Lee, Myung Jin Son, Sin-Hyoung Hong, Jae-Sung Ryu, Ji-Hyeon Min and Dong-Eon Lee; Visualization: Minji Lee, Myung Jin Son and Byeong-Rak Keum; Formal analysis: Minji Lee, Myung Jin Son, Ji Hoon Lee, Nam Doo Kim, Shi-Young Park, Darong Kim and Jeongmin Joo, Youngae Jung; Resources: Ji Hoon Lee, Nam Doo Kim, Darong Kim and Jeongmin Joo; Funding acquisition: Gun-Hwa Kim; Project administration: Koon Soon Kim and Gun-Hwa Kim; Supervision: Gun-Hwa Kim; Writing—original draft: Minji Lee; Writing—review & editing: Gun-Hwa Kim. All authors have read and approved the article.
The authors declare no conflict of interest.
This work was supported by grants from the Korea Basic Science Institute (Grant number C270300) and the National Research Foundation of Korea (Grant number NRF-2021R1A2C1008663).
期刊介绍:
Clinical and Translational Medicine (CTM) is an international, peer-reviewed, open-access journal dedicated to accelerating the translation of preclinical research into clinical applications and fostering communication between basic and clinical scientists. It highlights the clinical potential and application of various fields including biotechnologies, biomaterials, bioengineering, biomarkers, molecular medicine, omics science, bioinformatics, immunology, molecular imaging, drug discovery, regulation, and health policy. With a focus on the bench-to-bedside approach, CTM prioritizes studies and clinical observations that generate hypotheses relevant to patients and diseases, guiding investigations in cellular and molecular medicine. The journal encourages submissions from clinicians, researchers, policymakers, and industry professionals.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


