Findings from the individualized management of a patient with Acyl-CoA Oxidase-1 (ACOX1) deficiency: A bedside-to-bench-to-bedside strategy
IF 3.7
2区 生物学
Q2 ENDOCRINOLOGY & METABOLISM
引用次数: 0
Abstract
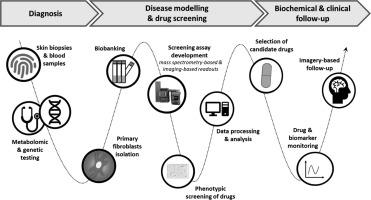
Acyl-CoA Oxidase-1 (ACOX1) deficiency (MIM 264470) is an autosomal recessive disease characterized by impairments in the desaturation of acyl-CoAs to 2-trans-enoyl-CoAs, which is the first step in the catalysis of the β-oxidative breakdown of very long chain fatty acids (VLCFA) occuring in peroxisomes. The deleterious accumulation of VLCFA in several organs, including the brain, is a key biochemical feature of this disease which has devastating neurological consequences. ACOX1 deficiency is ultra-rare; as such, few studies have been conducted to determine the leading causes of symptoms or uncover new therapeutics. When confronted with one such case, we decided to bring drug discovery tools to the patient's bedside in an attempt to identify a cure. A skin biopsy was performed on a young patient with ACOX1 deficiency, following which screening technologies and mass spectrometry analysis techniques were applied to design a cellular assay that enabled the direct measurement of the effect of small molecules on the patient's primary fibroblasts. This approach is particularly well adapted to inherited metabolic disorders such as ACOX1 deficiency. Through the evaluation of a proprietary library of repurposable drugs, we found that the anthelmintic drug niclosamide led to a significant reduction in VLCFA in vitro. This drug was subsequently administered to the patient for more than six years. This study outlines the screening and drug selection processes. Additionally, we present our comprehensive clinical and biochemical findings that aided in understanding the patient's natural history and analysis of the progression of the patient's symptoms throughout the treatment period. Although the patient's overall lifespan was extended compared to the average age at death in severe early onset cases of ACOX1 deficiency, we did not observe any definitive evidence of clinical or biochemical improvement during niclosamide treatment. Nonetheless, our study shows a good safety profile of long-term niclosamide administration in a child with a rare neurodegenerative disease, and illustrates the potential of individualized therapeutic strategies in the management of inherited metabolic disorders, which could benefit both patients and the broader scientific and medical communities.
对乙酰辅酶氧化酶-1 (ACOX1)缺乏症患者进行个体化管理的发现:从床边到床头再到床边的策略。
酰基-CoA 氧化酶-1(ACOX1)缺乏症(MIM 264470)是一种常染色体隐性遗传病,其特征是酰基-CoAs 向 2-反式-烯酰-CoAs 的脱饱和过程出现障碍,而这是过氧物酶体中发生的超长链脂肪酸(VLCFA)β-氧化分解催化过程的第一步。VLCFA 在包括大脑在内的多个器官中的有害积累是这种疾病的一个关键生化特征,它对神经系统造成了破坏性后果。ACOX1 缺乏症极为罕见,因此很少有研究能确定症状的主要原因或发现新疗法。面对这样一个病例,我们决定将药物发现工具带到病人的床边,试图找出治疗方法。我们对一名患有 ACOX1 缺乏症的年轻患者进行了皮肤活组织检查,随后应用筛选技术和质谱分析技术设计了一种细胞检测方法,能够直接测量小分子对患者原发性成纤维细胞的影响。这种方法尤其适用于 ACOX1 缺乏症等遗传代谢性疾病。通过对可再利用的专有药物库进行评估,我们发现抗蠕虫药物尼可刹米在体外可显著降低 VLCFA。随后,我们对该患者进行了长达六年多的药物治疗。本研究概述了筛选和药物选择过程。此外,我们还介绍了全面的临床和生化研究结果,这些结果有助于了解患者的自然病史,并分析了整个治疗期间患者症状的进展情况。虽然与严重的 ACOX1 缺乏症早发病例的平均死亡年龄相比,患者的总体寿命有所延长,但在尼可刹米的治疗过程中,我们并未观察到任何临床或生化改善的确切证据。尽管如此,我们的研究表明,对一名患有罕见神经退行性疾病的儿童长期服用尼可刹米具有良好的安全性,并说明了个体化治疗策略在治疗遗传性代谢紊乱方面的潜力,这将使患者和更广泛的科学界和医学界受益。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Molecular genetics and metabolism
生物-生化与分子生物学
CiteScore
5.90
自引率
7.90%
发文量
621
审稿时长
34 days
期刊介绍:
Molecular Genetics and Metabolism contributes to the understanding of the metabolic and molecular basis of disease. This peer reviewed journal publishes articles describing investigations that use the tools of biochemical genetics and molecular genetics for studies of normal and disease states in humans and animal models.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


