Jakob Wernig, Stefan Pilz, Christian Trummer, Verena Theiler-Schwetz, Lisa Maria Schmitt, Oleksiy Tsybrovskyy
{"title":"Challenging Diagnostic Workup of a 22-year-old Patient With Primary Pigmented Nodular Adrenocortical Disease.","authors":"Jakob Wernig, Stefan Pilz, Christian Trummer, Verena Theiler-Schwetz, Lisa Maria Schmitt, Oleksiy Tsybrovskyy","doi":"10.1210/jcemcr/luae174","DOIUrl":null,"url":null,"abstract":"<p><p>Primary pigmented nodular adrenocortical disease (PPNAD) is a rare cause of ACTH-independent Cushing syndrome (CS), presenting diagnostic challenges due to its rarity and its difficult clinical differentiation from other causes of CS. Here, we report the case of a 22-year-old female who developed classical symptoms of hypercortisolism including progressive weight gain, moon facies, and various skin manifestations. Despite biochemical screening confirming ACTH-independent CS, imaging modalities including computed tomography and magnetic resonance imaging showed normal adrenal gland morphology, complicating the localization of cortisol hypersecretion. Subsequent nuclear imaging methods were not indicative of ectopic cortisol production until adrenal vein sampling (AVS) conclusively identified the adrenal glands as the only possible source of cortisol hypersecretion. Eventually, bilateral adrenalectomy led to a significant improvement in symptoms. Pathological examination confirmed the diagnosis of PPNAD, and genetic testing revealed a mutation in the <i>PRKAR1A</i> gene associated with the Carney complex. This case highlights the importance of considering rare etiologies in hypercortisolism diagnosis and describes their challenging diagnostic workup and the utility of AVS in localizing cortisol hypersecretion in PPNAD patients.</p>","PeriodicalId":73540,"journal":{"name":"JCEM case reports","volume":"2 10","pages":"luae174"},"PeriodicalIF":0.0000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11443333/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JCEM case reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1210/jcemcr/luae174","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
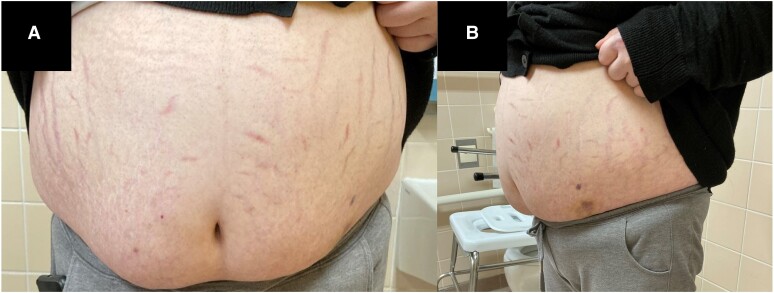
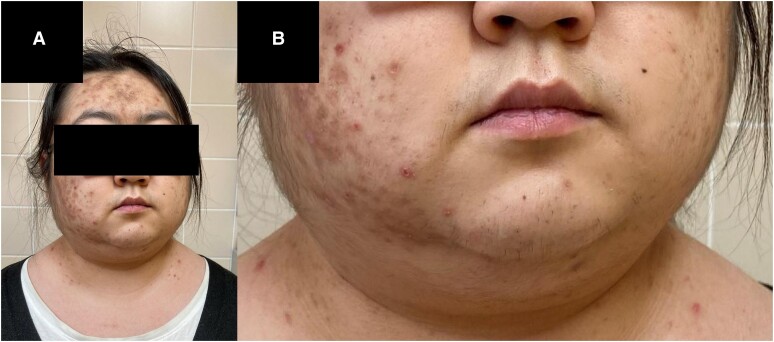
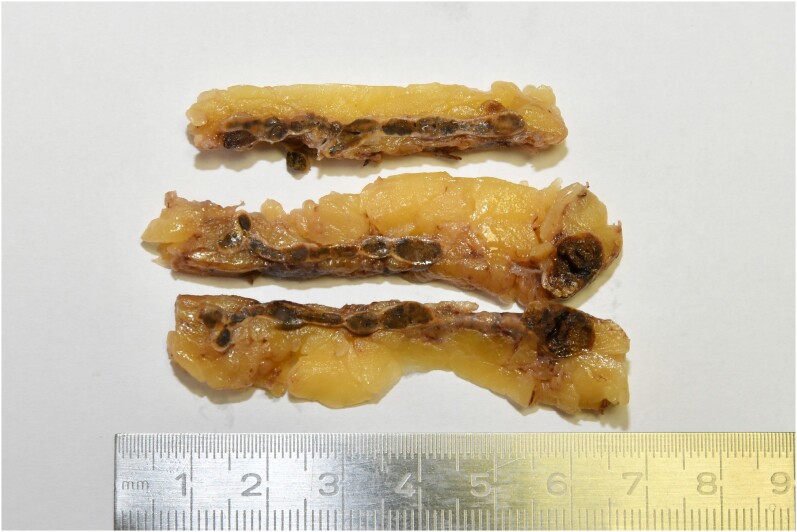
Primary pigmented nodular adrenocortical disease (PPNAD) is a rare cause of ACTH-independent Cushing syndrome (CS), presenting diagnostic challenges due to its rarity and its difficult clinical differentiation from other causes of CS. Here, we report the case of a 22-year-old female who developed classical symptoms of hypercortisolism including progressive weight gain, moon facies, and various skin manifestations. Despite biochemical screening confirming ACTH-independent CS, imaging modalities including computed tomography and magnetic resonance imaging showed normal adrenal gland morphology, complicating the localization of cortisol hypersecretion. Subsequent nuclear imaging methods were not indicative of ectopic cortisol production until adrenal vein sampling (AVS) conclusively identified the adrenal glands as the only possible source of cortisol hypersecretion. Eventually, bilateral adrenalectomy led to a significant improvement in symptoms. Pathological examination confirmed the diagnosis of PPNAD, and genetic testing revealed a mutation in the PRKAR1A gene associated with the Carney complex. This case highlights the importance of considering rare etiologies in hypercortisolism diagnosis and describes their challenging diagnostic workup and the utility of AVS in localizing cortisol hypersecretion in PPNAD patients.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


