Ab initio molecular dynamics simulations on the combustion mechanism of Al/Fe2O3 nanothermite at various temperatures
IF 3.1
3区 材料科学
Q2 MATERIALS SCIENCE, MULTIDISCIPLINARY
引用次数: 0
Abstract
Acquiring a comprehensive understanding of the combustion mechanism of nanothermites is crucial for elucidating phenomena and enhancing performance. The ab initio molecular dynamics method was employed to investigate the reaction process of the Al/Fe2O3 nanothermite at various temperatures. The complex combustion behavior and reaction mechanism were qualitatively described in terms of dynamic morphologies, potential energy, atomic density distribution, etc. Results suggest the initial reaction of Al/Fe2O3 is initiated by the migration of interfacial O atoms and the dissociation of Fe-O bonds, subsequently leading to the formation of alumina at the interface, which impedes the further progression of the thermite reaction. The increase in temperature enhances atomic diffusion and provides sufficient energy for the reaction. The interfacial metal Al layer undergoes melting and diffuses into the iron oxide layer, while vacancies generated during the reaction process sustain the continuous migration of internal oxygen atoms. At 2000 K and 3200 K, the initial structures completely collapse, facilitating the inward propagation of the thermite reaction, which subsequently results in the formation of alumina, iron clusters, and intermetallic compounds (AlFe, AlFe3, and Al6Fe). These findings offer significant insights into the combustion reaction mechanisms of nanothermites.
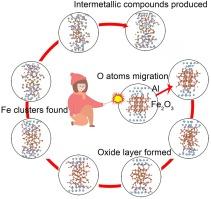
不同温度下 Al/Fe2O3 纳米热敏电阻燃烧机理的 Ab initio 分子动力学模拟
全面了解纳米热物的燃烧机理对于阐明现象和提高性能至关重要。本文采用ab initio分子动力学方法研究了Al/Fe2O3纳米热物在不同温度下的反应过程。从动态形貌、势能、原子密度分布等方面对复杂的燃烧行为和反应机理进行了定性描述。结果表明,Al/Fe2O3 的初始反应是由界面 O 原子的迁移和 Fe-O 键的解离引发的,随后在界面上形成氧化铝,阻碍了热敏反应的进一步进行。温度的升高加强了原子扩散,为反应提供了足够的能量。界面金属铝层发生熔化并扩散到氧化铁层中,而反应过程中产生的空位则维持着内部氧原子的不断迁移。在 2000 K 和 3200 K 时,初始结构完全坍塌,促进了热敏反应的向内扩展,随后形成氧化铝、铁簇和金属间化合物(AlFe、AlFe3 和 Al6Fe)。这些发现为了解纳米热敏物质的燃烧反应机制提供了重要启示。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational Materials Science
工程技术-材料科学:综合
CiteScore
6.50
自引率
6.10%
发文量
665
审稿时长
26 days
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


