{"title":"Transition from synchronous to asynchronous mechanisms in 1,3-dipolar cycloadditions: a polarizability perspective","authors":"César Barrales-Martínez, Rocío Durán, Pablo Jaque","doi":"10.1007/s00894-024-06161-2","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>This study investigates the energetic and polarizability characteristics of three 1,3-dipolar cycloaddition reactions between diazene oxide and substituted ethylenes, focusing on the transition from synchronous to asynchronous mechanisms. Synchronicity analysis, using the reaction force constant, indicates that the bond evolution process becomes increasingly decoupled as the number of cyano groups increases. Polarizability analysis reveals that isotropic polarizability reaches its maximum near the transition state in all cases, while anisotropy of polarizability shifts from the transition state toward the product direction as asynchronicity increases. The larger the shift, the more asynchronous the mechanism, as reflected by the weight of the transition region. A detailed examination of the parallel and perpendicular polarizability components to the newly formed sigma bonds shows that the evolution of the parallel component is closely aligned with the energetic changes along the reaction coordinate, particularly in the synchronous reaction. We have also identified a relationship between the displacement in the maximum state of the parallel component from the transition state and the synchronicity of the mechanism. The larger the displacement, the more asynchronous the mechanism. These findings suggest that asynchronous 1,3-dipolar cycloaddition mechanisms are characterized by a decoupling of isotropic and anisotropic polarizabilities and a shift in the maximum polarizability state of the parallel component toward the product direction.</p><h3>Methods</h3><p>Density functional theory calculations were performed at the B3LYP/6–311 + + G(d,p)//B3LYP/6-31G(d,p) level of theory. The polarizability was calculated at each point of the reaction path, obtained using the intrinsic reaction coordinate method, as implemented in Gaussian 16.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 10","pages":""},"PeriodicalIF":2.1000,"publicationDate":"2024-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06161-2","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
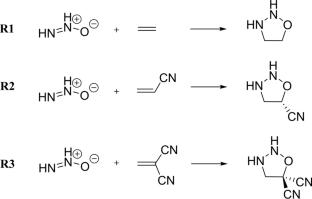
This study investigates the energetic and polarizability characteristics of three 1,3-dipolar cycloaddition reactions between diazene oxide and substituted ethylenes, focusing on the transition from synchronous to asynchronous mechanisms. Synchronicity analysis, using the reaction force constant, indicates that the bond evolution process becomes increasingly decoupled as the number of cyano groups increases. Polarizability analysis reveals that isotropic polarizability reaches its maximum near the transition state in all cases, while anisotropy of polarizability shifts from the transition state toward the product direction as asynchronicity increases. The larger the shift, the more asynchronous the mechanism, as reflected by the weight of the transition region. A detailed examination of the parallel and perpendicular polarizability components to the newly formed sigma bonds shows that the evolution of the parallel component is closely aligned with the energetic changes along the reaction coordinate, particularly in the synchronous reaction. We have also identified a relationship between the displacement in the maximum state of the parallel component from the transition state and the synchronicity of the mechanism. The larger the displacement, the more asynchronous the mechanism. These findings suggest that asynchronous 1,3-dipolar cycloaddition mechanisms are characterized by a decoupling of isotropic and anisotropic polarizabilities and a shift in the maximum polarizability state of the parallel component toward the product direction.
Methods
Density functional theory calculations were performed at the B3LYP/6–311 + + G(d,p)//B3LYP/6-31G(d,p) level of theory. The polarizability was calculated at each point of the reaction path, obtained using the intrinsic reaction coordinate method, as implemented in Gaussian 16.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


