Traversing chemical space with active deep learning for low-data drug discovery
IF 12
Q1 COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS
引用次数: 0
Abstract
Deep learning is accelerating drug discovery. However, current approaches are often affected by limitations in the available data, in terms of either size or molecular diversity. Active deep learning has high potential for low-data drug discovery, as it allows iterative model improvement during the screening process. However, there are several ‘known unknowns’ that limit the wider adoption of active deep learning in drug discovery: (1) what the best computational strategies are for chemical space exploration, (2) how active learning holds up to traditional, non-iterative, approaches and (3) how it should be used in the low-data scenarios typical of drug discovery. To provide answers, this study simulates a low-data drug discovery scenario, and systematically analyzes six active learning strategies combined with two deep learning architectures, on three large-scale molecular libraries. We identify the most important determinants of success in low-data regimes and show that active learning can achieve up to a sixfold improvement in hit discovery when compared with traditional screening methods. Active deep learning is a promising approach to learn from low-data scenarios in drug discovery. This study illuminates key success factors of active learning and shows that it can boost hit discovery by up to sixfold over traditional methods.
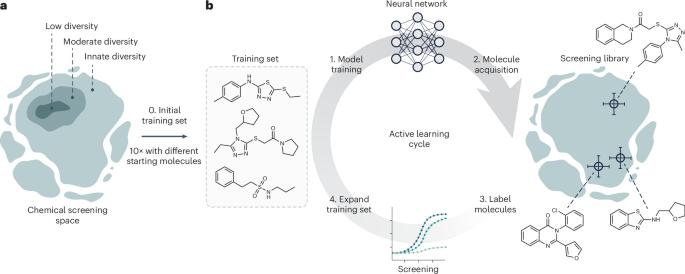
利用主动深度学习穿越化学空间,实现低数据药物发现。
深度学习正在加速药物发现。然而,目前的方法往往受到可用数据规模或分子多样性的限制。主动深度学习在低数据药物发现方面具有很大潜力,因为它允许在筛选过程中迭代改进模型。然而,有几个 "已知的未知数 "限制了主动深度学习在药物发现中的广泛应用:(1)化学空间探索的最佳计算策略是什么;(2)主动学习与传统的非迭代方法相比有何优势;(3)在药物发现的典型低数据场景中应如何使用主动学习。为了提供答案,本研究模拟了低数据药物发现场景,并在三个大规模分子库上系统分析了六种主动学习策略与两种深度学习架构的结合。我们确定了在低数据环境中取得成功的最重要决定因素,并表明与传统筛选方法相比,主动学习可以在发现新药方面实现高达六倍的改进。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


