Revealing of Intrinsic Relation between Surface Atomic Geometry and Catalytic Mechanism of CuNi Alloy Catalyst in CO2 Hydrogenation to Methanol
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
It is well-known that the catalytic mechanism and performance are strongly dependent on the exposed surfaces of metal catalysts. This concept is further broadened and elaborated in the current work for the bimetallic NiCu catalyst in CO2 hydrogenation to methanol. (100), (110), and (111) facets of NiCu with either Cu or Ni termination, which consisted of five different surfaces, are investigated by DFT-based microkinetic simulation. The unique surface geometry of the alloy surface results in the distinct electronic structure as revealed from electron localization function and PDOS analyses. CO2 molecule exhibits strong chemisorption on all surfaces with a bent bond angle which is significantly activated upon adsorption. Three mechanisms, including formate, RWGS + COhydro, and CO + O, are explored at the same footing. It is concluded that the first step in the pathway has pivotal importance to determine the favorable mechanism. From the reaction pathway analysis, it is found that direct C–O bond breaking has a comparable barrier with the hydrogenation step which is not usually observed for metallic catalysts in CO2 hydrogenation. In general, hydrogen addition to carbon in the CO2 molecule has a smaller barrier than the counterpart of addition to oxygen. The precisely exposed atom on the bimetallic catalyst largely determined both performance and mechanism which complemented the concept of surface-dependent catalytic properties. Microkinetic simulation indicated that the (111) surface with Cu termination has the largest TOF of methanol formation in the experimental relevant temperature, and the favorable mechanism is ascribed to be formate. Moreover, both thermodynamics and kinetic control steps are identified from the degree of rate control analysis. Overall, the current work deepened the importance of fine tuning the surface geometry to adjust the catalytic performance and mechanism and paved the way for further development of bimetallic catalysts for CO2 hydrogenation.
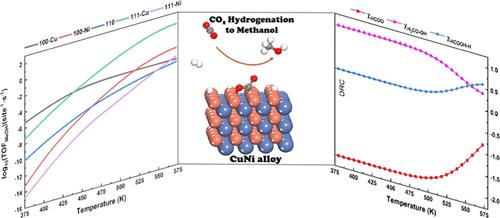
揭示 CuNi 合金催化剂在 CO2 加氢制甲醇过程中的表面原子几何与催化机理之间的内在联系
众所周知,催化机理和性能在很大程度上取决于金属催化剂的暴露表面。本研究针对二氧化碳加氢制甲醇的双金属镍铜催化剂进一步拓宽和阐述了这一概念。通过基于 DFT 的微动力学模拟,研究了以铜或镍为端点的 NiCu 的 (100)、(110) 和 (111) 面,它们由五个不同的表面组成。电子局域函数和 PDOS 分析表明,合金表面独特的几何形状产生了独特的电子结构。二氧化碳分子在所有表面上都表现出强烈的化学吸附作用,其弯曲的键角在吸附后被显著激活。在同一基础上探讨了三种机制,包括甲酸盐、RWGS + COhydro 和 CO + O。结论是,路径中的第一步对于确定有利的机理至关重要。从反应路径分析中发现,C-O 键的直接断裂与氢化步骤具有相当的障碍,这在二氧化碳氢化的金属催化剂中并不常见。一般来说,氢与 CO2 分子中碳的加成反应比与氧的加成反应的障碍要小。双金属催化剂上精确暴露的原子在很大程度上决定了催化剂的性能和机理,补充了表面催化特性的概念。微动力学模拟表明,在实验相关温度下,以 Cu 为末端的 (111) 表面形成甲醇的 TOF 最大,其有利机制被归结为甲酸盐。此外,从速率控制分析中还发现了热力学和动力学控制步骤。总之,本研究深化了微调表面几何形状对调整催化性能和机理的重要性,为进一步开发二氧化碳加氢双金属催化剂铺平了道路。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


