Yiming Qin, Zihan Chen, Ye Peng, Ying Xiao, Tian Zhong, Xi Yu
{"title":"Deep learning methods for protein structure prediction","authors":"Yiming Qin, Zihan Chen, Ye Peng, Ying Xiao, Tian Zhong, Xi Yu","doi":"10.1002/mef2.96","DOIUrl":null,"url":null,"abstract":"<p>Protein structure prediction (PSP) has been a prominent topic in bioinformatics and computational biology, aiming to predict protein function and structure from sequence data. The three-dimensional conformation of proteins is pivotal for their intricate biological roles. With the advancement of computational capabilities and the adoption of deep learning (DL) technologies (especially Transformer network architectures), the PSP field has ushered in a brand-new era of “neuralization.” Here, we focus on reviewing the evolution of PSP from traditional to modern deep learning-based approaches and the characteristics of various structural prediction methods. This emphasizes the advantages of deep learning-based hybrid prediction methods over traditional approaches. This study also provides a summary analysis of widely used bioinformatics databases and the latest structure prediction models. It discusses deep learning networks and algorithmic optimization for model training, validation, and evaluation. In addition, a summary discussion of the major advances in deep learning-based protein structure prediction is presented. The update of AlphaFold 3 further extends the boundaries of prediction models, especially in protein-small molecule structure prediction. This marks a key shift toward a holistic approach in biomolecular structure elucidation, aiming at solving almost all sequence-to-structure puzzles in various biological phenomena.</p>","PeriodicalId":74135,"journal":{"name":"MedComm - Future medicine","volume":"3 3","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mef2.96","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"MedComm - Future medicine","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mef2.96","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
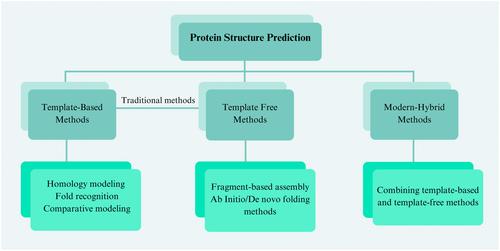
Protein structure prediction (PSP) has been a prominent topic in bioinformatics and computational biology, aiming to predict protein function and structure from sequence data. The three-dimensional conformation of proteins is pivotal for their intricate biological roles. With the advancement of computational capabilities and the adoption of deep learning (DL) technologies (especially Transformer network architectures), the PSP field has ushered in a brand-new era of “neuralization.” Here, we focus on reviewing the evolution of PSP from traditional to modern deep learning-based approaches and the characteristics of various structural prediction methods. This emphasizes the advantages of deep learning-based hybrid prediction methods over traditional approaches. This study also provides a summary analysis of widely used bioinformatics databases and the latest structure prediction models. It discusses deep learning networks and algorithmic optimization for model training, validation, and evaluation. In addition, a summary discussion of the major advances in deep learning-based protein structure prediction is presented. The update of AlphaFold 3 further extends the boundaries of prediction models, especially in protein-small molecule structure prediction. This marks a key shift toward a holistic approach in biomolecular structure elucidation, aiming at solving almost all sequence-to-structure puzzles in various biological phenomena.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


