Using ab initio molecular dynamics to determine the mobility of single, di and tri self-interstitial configurations in hcp Zr
IF 5.3
2区 材料科学
Q2 MATERIALS SCIENCE, MULTIDISCIPLINARY
引用次数: 0
Abstract
Atomic configurations and activation energies for diffusion of point defects and point defects clusters are important elements to consider when predicting the microstructural evolution of zirconium-based materials under irradiation. Using Density Functional Theory (DFT) we performed molecular dynamics (MD) calculations to determine the diffusion coefficient of self-interstitials (SIA) clusters containing up to three atoms in the hexagonal close packed (hcp) structure of zirconium. The calculated values of the activation energy for single SIA diffusion are in line with previous DFT studies performed with accurate constrained minimization techniques, which validates the adopted methodology. Diffusion in the basal plane is similar for mono-, di- and tri-SIAs but diffusion along the axis is significantly slowed down by the addition of SIA. Quantitative differences with existing empirical potential results highlight the usefulness of the ab initio MD calculations reported in this work.
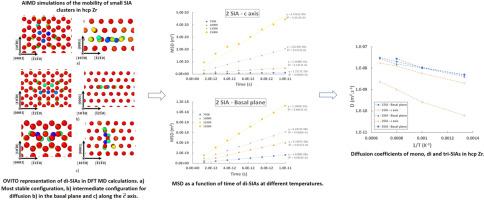
利用 ab initio 分子动力学确定 hcp Zr 中单、二和三自间隙构型的迁移率
在预测辐照下锆基材料的微观结构演变时,点缺陷和点缺陷簇扩散的原子构型和活化能是需要考虑的重要因素。我们利用密度泛函理论(DFT)进行了分子动力学(MD)计算,以确定在锆的六方紧密堆积(hcp)结构中最多包含三个原子的自间隙(SIA)簇的扩散系数。单个 SIA 扩散的活化能计算值与之前使用精确约束最小化技术进行的 DFT 研究结果一致,这验证了所采用的方法。单SIA、二SIA和三SIA在基底面上的扩散相似,但沿c→轴的扩散因加入SIA而明显减慢。与现有经验电位结果的定量差异凸显了本研究中报告的 ab initio MD 计算的实用性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Scripta Materialia
工程技术-材料科学:综合
CiteScore
11.40
自引率
5.00%
发文量
581
审稿时长
34 days
期刊介绍:
Scripta Materialia is a LETTERS journal of Acta Materialia, providing a forum for the rapid publication of short communications on the relationship between the structure and the properties of inorganic materials. The emphasis is on originality rather than incremental research. Short reports on the development of materials with novel or substantially improved properties are also welcomed. Emphasis is on either the functional or mechanical behavior of metals, ceramics and semiconductors at all length scales.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


