Clinical Assessment of Breast Cancer Resistance Protein (BCRP)-Mediated Drug-Drug Interactions of Sepiapterin with Curcumin and Rosuvastatin in Healthy Volunteers.
Lan Gao, Diksha Kaushik, Kimberly Ingalls, Sarah Milner, Neil Smith, Ronald Kong
{"title":"Clinical Assessment of Breast Cancer Resistance Protein (BCRP)-Mediated Drug-Drug Interactions of Sepiapterin with Curcumin and Rosuvastatin in Healthy Volunteers.","authors":"Lan Gao, Diksha Kaushik, Kimberly Ingalls, Sarah Milner, Neil Smith, Ronald Kong","doi":"10.1007/s40268-024-00488-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objective: </strong>Sepiapterin, also known as PTC923 and CNSA-001, is a synthetic form of endogenous sepiapterin being developed as a novel oral treatment for phenylketonuria. Sepiapterin is a natural precursor of tetrahydrobiopterin (BH<sub>4</sub>) and, when orally administered, is converted to BH<sub>4</sub> via the pterin salvage pathway. In vitro studies have demonstrated that both sepiapterin and BH<sub>4</sub> are both substrates and inhibitors of the breast cancer resistance protein (BCRP) transporter. This phase I study investigated BCRP-mediated drug-drug interactions of sepiapterin as a victim and as a perpetrator.</p><p><strong>Methods: </strong>An open-label, fixed-sequence, four-period, crossover, single-dose study was conducted in adult male and female healthy volunteers (18-55 years of age). In a given treatment period, subjects received a single oral dose of sepiapterin (20 mg/kg), sepiapterin (20 mg/kg) plus curcumin (2 g), rosuvastatin (10 mg), or rosuvastatin (10 mg) plus sepiapterin (60 mg/kg). The pharmacokinetics of sepiapterin, its metabolite BH<sub>4</sub>, and rosuvastatin were studied, and geometric mean ratios of exposures in the presence and absence of the BCRP inhibitor curcumin or sepiapterin were estimated. The presence of the BCRP c.421C>A polymorphism was evaluated in all subjects.</p><p><strong>Results: </strong>A total of 29 subjects were enrolled and included in the safety analysis. Among them, 26 subjects were included in the pharmacokinetic and drug-drug interaction analyses. Following oral administration 20 mg/kg sepiapterin, sepiapterin was rapidly and extensively converted to BH<sub>4</sub>, and BH<sub>4</sub> maximum observed concentration (415.0 ng/mL) was observed 4.95 h (time to maximum observed concentration) post-dose. Sepiapterin maximum observed concentration and area under the concentration-time curve from time 0 to time of the last quantifiable measurement or the last sample collection time (AUC<sub>last</sub>) were <1% of BH<sub>4</sub> values. Coadministration of the BCRP inhibitor curcumin (2 g) increased BH<sub>4</sub> maximum observed concentration, AUC<sub>last</sub>, and area under the concentration-time curve from time 0 extrapolated to infinity by 24%, 21%, and 20%, respectively. When sepiapterin was coadministered with the BCRP substrate rosuvastatin, there was no effect on the pharmacokinetics of rosuvastatin. BCRP c.421C/A carriers (n = 4) had higher plasma exposures of BH<sub>4</sub> (1.39 × for AUC<sub>last</sub>) and rosuvastatin (1.61 × for AUC<sub>last</sub>) than c.421C/C carriers (n = 22). Greater increases in BH<sub>4</sub> exposures (1.33 vs 1.18 for AUC<sub>last</sub>) were observed in c.421C/A carriers compared with c.421C/C carriers when sepiapterin was coadministered with curcumin. All treatments were well tolerated during the study.</p><p><strong>Conclusions: </strong>Oral coadministration of the BCRP inhibitor curcumin slightly increased the plasma exposure of sepiapterin and its metabolite BH<sub>4</sub> in healthy volunteers. This modest increase was deemed not clinically meaningful. Sepiapterin did not alter the pharmacokinetics of the BCRP substrate rosuvastatin.</p>","PeriodicalId":49258,"journal":{"name":"Drugs in Research & Development","volume":" ","pages":"477-487"},"PeriodicalIF":2.1000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11455768/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Drugs in Research & Development","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s40268-024-00488-0","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/24 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
Background and objective: Sepiapterin, also known as PTC923 and CNSA-001, is a synthetic form of endogenous sepiapterin being developed as a novel oral treatment for phenylketonuria. Sepiapterin is a natural precursor of tetrahydrobiopterin (BH4) and, when orally administered, is converted to BH4 via the pterin salvage pathway. In vitro studies have demonstrated that both sepiapterin and BH4 are both substrates and inhibitors of the breast cancer resistance protein (BCRP) transporter. This phase I study investigated BCRP-mediated drug-drug interactions of sepiapterin as a victim and as a perpetrator.
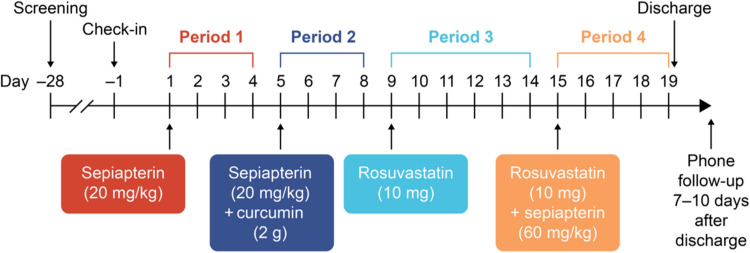
Methods: An open-label, fixed-sequence, four-period, crossover, single-dose study was conducted in adult male and female healthy volunteers (18-55 years of age). In a given treatment period, subjects received a single oral dose of sepiapterin (20 mg/kg), sepiapterin (20 mg/kg) plus curcumin (2 g), rosuvastatin (10 mg), or rosuvastatin (10 mg) plus sepiapterin (60 mg/kg). The pharmacokinetics of sepiapterin, its metabolite BH4, and rosuvastatin were studied, and geometric mean ratios of exposures in the presence and absence of the BCRP inhibitor curcumin or sepiapterin were estimated. The presence of the BCRP c.421C>A polymorphism was evaluated in all subjects.
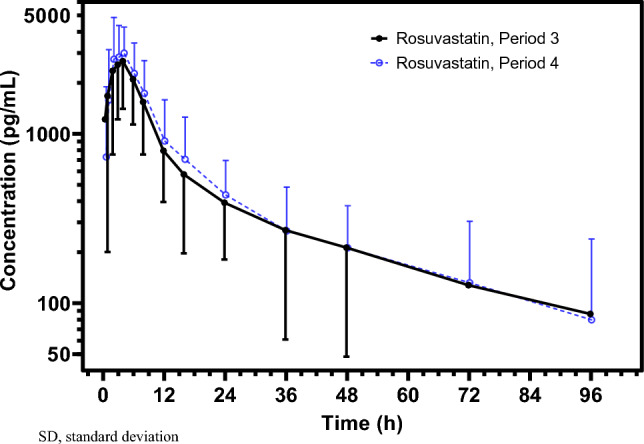
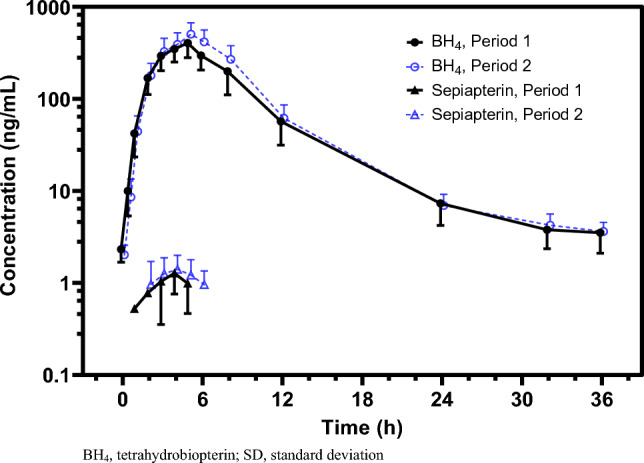
Results: A total of 29 subjects were enrolled and included in the safety analysis. Among them, 26 subjects were included in the pharmacokinetic and drug-drug interaction analyses. Following oral administration 20 mg/kg sepiapterin, sepiapterin was rapidly and extensively converted to BH4, and BH4 maximum observed concentration (415.0 ng/mL) was observed 4.95 h (time to maximum observed concentration) post-dose. Sepiapterin maximum observed concentration and area under the concentration-time curve from time 0 to time of the last quantifiable measurement or the last sample collection time (AUClast) were <1% of BH4 values. Coadministration of the BCRP inhibitor curcumin (2 g) increased BH4 maximum observed concentration, AUClast, and area under the concentration-time curve from time 0 extrapolated to infinity by 24%, 21%, and 20%, respectively. When sepiapterin was coadministered with the BCRP substrate rosuvastatin, there was no effect on the pharmacokinetics of rosuvastatin. BCRP c.421C/A carriers (n = 4) had higher plasma exposures of BH4 (1.39 × for AUClast) and rosuvastatin (1.61 × for AUClast) than c.421C/C carriers (n = 22). Greater increases in BH4 exposures (1.33 vs 1.18 for AUClast) were observed in c.421C/A carriers compared with c.421C/C carriers when sepiapterin was coadministered with curcumin. All treatments were well tolerated during the study.
Conclusions: Oral coadministration of the BCRP inhibitor curcumin slightly increased the plasma exposure of sepiapterin and its metabolite BH4 in healthy volunteers. This modest increase was deemed not clinically meaningful. Sepiapterin did not alter the pharmacokinetics of the BCRP substrate rosuvastatin.
期刊介绍:
Drugs in R&D is an international, peer reviewed, open access, online only journal, and provides timely information from all phases of drug research and development that will inform clinical practice. Healthcare decision makers are thus provided with knowledge about the developing place of a drug in therapy.
The Journal includes:
Clinical research on new and established drugs;
Preclinical research of direct relevance to clinical drug development;
Short communications and case study reports that meet the above criteria will also be considered;
Reviews may also be considered.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


