Hederagenin inhibits mitochondrial damage in Parkinson’s disease via mitophagy induction
IF 7.1
2区 生物学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
Background
Parkinson's disease (PD) is a neurodegenerative disorder marked by the loss of dopaminergic neurons and the formation of α-synuclein aggregates. Mitochondrial dysfunction and oxidative stress are pivotal in PD pathogenesis, with impaired mitophagy contributing to the accumulation of mitochondrial damage. Hederagenin (Hed), a natural triterpenoid, has shown potential neuroprotective effects; however, its mechanisms of action in PD models are not fully understood.
Method
We investigated the effects of Hed on 6-hydroxydopamine (6-OHDA)-induced cytotoxicity in SH-SY5Y cells by assessing cell viability, mitochondrial function, and oxidative stress markers. Mitophagy induction was evaluated using autophagy and mitophagy inhibitors and fluorescent staining techniques. Additionally, transgenic Caenorhabditis elegans (C. elegans) models of PD were used to validate the neuroprotective effects of Hed in vivo by focusing on α-synuclein aggregation, mobility, and dopaminergic neuron integrity.
Results
Hed significantly enhanced cell viability in 6-OHDA-treated SH-SY5Y cells by inhibiting cell death and reducing oxidative stress. It ameliorated mitochondrial damage, evidenced by decreased mitochondrial superoxide production, restored membrane potential, and improved mitochondrial morphology. Hed also induced mitophagy, as shown by increased autophagosome formation and reduced oxidative stress; these effects were diminished by autophagy and mitophagy inhibitors. In C. elegans models, Hed activated mitophagy and reduced α-synuclein aggregation, improved mobility, and mitigated the loss of dopaminergic neurons. RNA interference targeting the mitophagy-related genes pdr-1 and pink-1 partially reversed these benefits, underscoring the role of mitophagy in Hed's neuroprotective actions.
Conclusion
Hed exhibits significant neuroprotective effects in both in vitro and in vivo PD models by enhancing mitophagy, reducing oxidative stress, and mitigating mitochondrial dysfunction. These findings suggest that Hed holds promise as a therapeutic agent for PD, offering new avenues for future research and potential drug development.
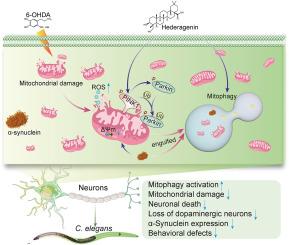
Hederagenin通过诱导有丝分裂抑制帕金森病的线粒体损伤。
背景:帕金森病(PD)是一种以多巴胺能神经元丧失和α-突触核蛋白聚集体形成为特征的神经退行性疾病。线粒体功能障碍和氧化应激在帕金森病的发病机制中起着关键作用,有丝分裂吞噬功能受损会导致线粒体损伤累积。Hederagenin(Hed)是一种天然三萜类化合物,具有潜在的神经保护作用,但其在帕金森病模型中的作用机制尚未完全明了:我们通过评估细胞活力、线粒体功能和氧化应激标记物,研究了Hed对6-羟基多巴胺(6-OHDA)诱导的SH-SY5Y细胞细胞毒性的影响。利用自噬和有丝分裂抑制剂以及荧光染色技术评估了有丝分裂的诱导作用。此外,研究人员还利用转基因秀丽隐杆线虫(C. elegans)模型,通过关注α-突触核蛋白的聚集、流动性和多巴胺能神经元的完整性,验证了赫德在体内的神经保护作用:结果:通过抑制细胞死亡和减少氧化应激,Hed能明显提高经6-OHDA处理的SH-SY5Y细胞的存活率。线粒体超氧化物生成的减少、膜电位的恢复和线粒体形态的改善表明,Hed 能改善线粒体损伤。Hed 还能诱导有丝分裂,表现为自噬体形成增加和氧化应激减少;自噬和有丝分裂抑制剂会减弱这些作用。在C.elegans模型中,Hed激活了有丝分裂,减少了α-突触核蛋白的聚集,提高了移动性,并减轻了多巴胺能神经元的损失。针对有丝分裂相关基因 pdr-1 和 pink-1 的 RNA 干扰部分逆转了这些益处,突出了有丝分裂在 Hed 神经保护作用中的作用:结论:通过增强有丝分裂、减少氧化应激和减轻线粒体功能障碍,Hed在体外和体内帕金森病模型中都表现出了明显的神经保护作用。这些研究结果表明,Hed有望成为一种治疗帕金森病的药物,为未来的研究和潜在药物开发提供了新的途径。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Free Radical Biology and Medicine
医学-内分泌学与代谢
CiteScore
14.00
自引率
4.10%
发文量
850
审稿时长
22 days
期刊介绍:
Free Radical Biology and Medicine is a leading journal in the field of redox biology, which is the study of the role of reactive oxygen species (ROS) and other oxidizing agents in biological systems. The journal serves as a premier forum for publishing innovative and groundbreaking research that explores the redox biology of health and disease, covering a wide range of topics and disciplines. Free Radical Biology and Medicine also commissions Special Issues that highlight recent advances in both basic and clinical research, with a particular emphasis on the mechanisms underlying altered metabolism and redox signaling. These Special Issues aim to provide a focused platform for the latest research in the field, fostering collaboration and knowledge exchange among researchers and clinicians.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


