João G. de Oliveira Neto, Jailton R. Viana, Kamila R. Abreu, Luiz F. L. da Silva, Mateus R. Lage, Stanislav R. Stoyanov, Francisco F. de Sousa, Rossano Lang, Adenilson O. dos Santos
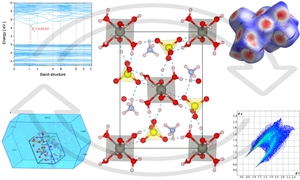
{"title":"Tutton salt (NH4)2Zn(SO4)2(H2O)6: thermostructural, spectroscopic, Hirshfeld surface, and DFT investigations","authors":"João G. de Oliveira Neto, Jailton R. Viana, Kamila R. Abreu, Luiz F. L. da Silva, Mateus R. Lage, Stanislav R. Stoyanov, Francisco F. de Sousa, Rossano Lang, Adenilson O. dos Santos","doi":"10.1007/s00894-024-06089-7","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Ammonium Tutton salts have been widely studied in recent years due to their thermostructural properties, which make them promising compounds for application in thermochemical energy storage devices. In this work, a detailed experimental study of the Tutton salt with the formula (NH<sub>4</sub>)<sub>2</sub>Zn(SO<sub>4</sub>)<sub>2</sub>(H<sub>2</sub>O)<sub>6</sub> is carried out. Its structural, vibrational, and thermal properties are analyzed and discussed. Powder X-ray diffraction (PXRD) studies confirm that the compound crystallizes in a structure of a Tutton salt, with monoclinic symmetry and <i>P</i>2<sub>1</sub>/<i>a</i> space group. The Hirshfeld surface analysis results indicate that the main contacts stabilizing the material crystal lattice are H···O/O···H, H···H, and O···O. In addition, a typical behavior of an insulating material is confirmed based on the electronic bandgap calculated from the band structure and experimental absorption coefficient. The Raman and infrared spectra calculated using DFT are in a good agreement with the respective experimental spectroscopic results. Thermal analysis in the range from 300 to 773 K reveals one exothermic and several endothermic events that are investigated using PXRD measurements as a function of temperature. With increasing temperature, two new structural phases are identified, one of which is resolved using the Le Bail method. Our findings suggest that the salt (NH<sub>4</sub>)<sub>2</sub>Zn(SO<sub>4</sub>)<sub>2</sub>(H<sub>2</sub>O)<sub>6</sub> is a promising thermochemical material suitable for the development of heat storage systems, due to its low dehydration temperature (≈ 330 K), high enthalpy of dehydration (122.43 kJ/mol of H<sub>2</sub>O), and hydration after 24 h.</p><h3>Methods</h3><p>Computational studies using Hirshfeld surfaces and void analysis are conducted to identify and quantify the intermolecular contacts occurring in the crystal structure. Furthermore, geometry optimization calculations are performed based on density functional theory (DFT) using the PBE functional and norm-conserving pseudopotentials implemented in the Cambridge Serial Total Energy Package (CASTEP). The primitive unit cell optimization was conducted using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm. The electronic properties of band structure and density of states, and vibrational modes of the optimized crystal lattice are calculated and analyzed.</p><h3>Graphical abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 10","pages":""},"PeriodicalIF":2.1000,"publicationDate":"2024-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s00894-024-06089-7.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06089-7","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
Ammonium Tutton salts have been widely studied in recent years due to their thermostructural properties, which make them promising compounds for application in thermochemical energy storage devices. In this work, a detailed experimental study of the Tutton salt with the formula (NH4)2Zn(SO4)2(H2O)6 is carried out. Its structural, vibrational, and thermal properties are analyzed and discussed. Powder X-ray diffraction (PXRD) studies confirm that the compound crystallizes in a structure of a Tutton salt, with monoclinic symmetry and P21/a space group. The Hirshfeld surface analysis results indicate that the main contacts stabilizing the material crystal lattice are H···O/O···H, H···H, and O···O. In addition, a typical behavior of an insulating material is confirmed based on the electronic bandgap calculated from the band structure and experimental absorption coefficient. The Raman and infrared spectra calculated using DFT are in a good agreement with the respective experimental spectroscopic results. Thermal analysis in the range from 300 to 773 K reveals one exothermic and several endothermic events that are investigated using PXRD measurements as a function of temperature. With increasing temperature, two new structural phases are identified, one of which is resolved using the Le Bail method. Our findings suggest that the salt (NH4)2Zn(SO4)2(H2O)6 is a promising thermochemical material suitable for the development of heat storage systems, due to its low dehydration temperature (≈ 330 K), high enthalpy of dehydration (122.43 kJ/mol of H2O), and hydration after 24 h.
Methods
Computational studies using Hirshfeld surfaces and void analysis are conducted to identify and quantify the intermolecular contacts occurring in the crystal structure. Furthermore, geometry optimization calculations are performed based on density functional theory (DFT) using the PBE functional and norm-conserving pseudopotentials implemented in the Cambridge Serial Total Energy Package (CASTEP). The primitive unit cell optimization was conducted using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm. The electronic properties of band structure and density of states, and vibrational modes of the optimized crystal lattice are calculated and analyzed.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


