Jia Fu, Meng Li, Chunying Rong, Dongbo Zhao, Shubin Liu
{"title":"Information-theoretic quantities as effective descriptors of electrophilicity and nucleophilicity in density functional theory","authors":"Jia Fu, Meng Li, Chunying Rong, Dongbo Zhao, Shubin Liu","doi":"10.1007/s00894-024-06116-7","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Electrophilicity and nucleophilicity are two vastly important chemical concepts gauging the capability of atoms in molecules to accept and donate the maximal number of electrons. In our earlier studies, we proposed to simultaneously quantify them using the Kullback–Leibler divergence from the information-theoretic approach in density functional theory. However, several issues with this scheme remain to be clarified such as its general validity, predictability, and relationship with other information-theoretic quantities. In this work, we revisit the matter with bigger datasets and deeper theoretical insights. Five information-theoretic quantities including Kullback–Leibler divergence, Hirshfeld charge, Ghost-Berkowitz-Parr entropy, and second and third orders of relative Onicescu information energy are found to be reliable and robust descriptors of electrophilicity and nucleophilicity propensities. Employing these five descriptors, we design a list of new compounds and predict their electrophilicity and nucleophilicity scales. This work should markedly improve our confidence and capability in applying information-theoretic quantities to evaluate electrophilicity and nucleophilicity propensities and henceforth pave the route for more applications of these quantities from information-theoretic approach in density functional theory in the future.</p><h3>Methods</h3><p>All structures were fully optimized at the M06-2X/6–311 + G(d) level of DFT functional using the Gaussian 16 package (version C01) with integration grids and tight self-consistent-field convergence. The solvent effect was taken into account by using the implicit solvent model (CPCM) in the CH<sub>2</sub>Cl<sub>2</sub> solvent, and all 3D contour surfaces of Fukui function, local temperature, and ITA (information-theoretic approach) quantities were generated by GaussView. The Multiwfn 3.8 program was used to calculate the ITA indexes and atomic charges.</p><h3>Graphical Abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 10","pages":""},"PeriodicalIF":2.1000,"publicationDate":"2024-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06116-7","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
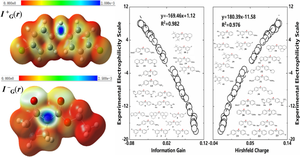
Electrophilicity and nucleophilicity are two vastly important chemical concepts gauging the capability of atoms in molecules to accept and donate the maximal number of electrons. In our earlier studies, we proposed to simultaneously quantify them using the Kullback–Leibler divergence from the information-theoretic approach in density functional theory. However, several issues with this scheme remain to be clarified such as its general validity, predictability, and relationship with other information-theoretic quantities. In this work, we revisit the matter with bigger datasets and deeper theoretical insights. Five information-theoretic quantities including Kullback–Leibler divergence, Hirshfeld charge, Ghost-Berkowitz-Parr entropy, and second and third orders of relative Onicescu information energy are found to be reliable and robust descriptors of electrophilicity and nucleophilicity propensities. Employing these five descriptors, we design a list of new compounds and predict their electrophilicity and nucleophilicity scales. This work should markedly improve our confidence and capability in applying information-theoretic quantities to evaluate electrophilicity and nucleophilicity propensities and henceforth pave the route for more applications of these quantities from information-theoretic approach in density functional theory in the future.
Methods
All structures were fully optimized at the M06-2X/6–311 + G(d) level of DFT functional using the Gaussian 16 package (version C01) with integration grids and tight self-consistent-field convergence. The solvent effect was taken into account by using the implicit solvent model (CPCM) in the CH2Cl2 solvent, and all 3D contour surfaces of Fukui function, local temperature, and ITA (information-theoretic approach) quantities were generated by GaussView. The Multiwfn 3.8 program was used to calculate the ITA indexes and atomic charges.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


