Selective degradation of mutant FMS-like tyrosine kinase-3 requires BIM-dependent depletion of heat shock proteins
IF 12.8
1区 医学
Q1 HEMATOLOGY
引用次数: 0
Abstract
Internal tandem duplications in the FMS-like tyrosine kinase-3 (FLT3-ITD) are common mutations in acute myeloid leukemia (AML). Proteolysis-targeting chimeras (PROTACs) that induce proteasomal degradation of mutated FLT3 emerge as innovative pharmacological approach. Molecular mechanisms that control targeted proteolysis beyond the ubiquitin-proteasome-system are undefined and PROTACs are the only known type of FLT3 degraders. We report that the von-Hippel-Lindau ubiquitin-ligase based FLT3 PROTAC MA49 (melotinib-49) and the FLT3 hydrophobic tagging molecule MA50 (halotinib-50) reduce endoplasmic reticulum-associated, oncogenic FLT3-ITD but spare FLT3. Nanomolar doses of MA49 and MA50 induce apoptosis of human leukemic cell lines and primary AML blasts with FLT3-ITD (p < 0.05-0.0001), but not of primary hematopoietic stem cells and differentiated immune cells, FLT3 wild-type cells, retinal cells, and c-KIT-dependent cells. In vivo activity of MA49 against FLT3-ITD-positive leukemia cells is verified in a Danio rerio model. The degrader-induced loss of FLT3-ITD involves the pro-apoptotic BH3-only protein BIM and a previously unidentified degrader-induced depletion of protein-folding chaperones. The expression levels of HSP90 and HSP110 correlate with reduced AML patient survival (p < 0.1) and HSP90, HSP110, and BIM are linked to the expression of FLT3 in primary AML cells (p < 0.01). HSP90 suppresses degrader-induced FLT3-ITD elimination and thereby establishes a mechanistically defined feed-back circuit.
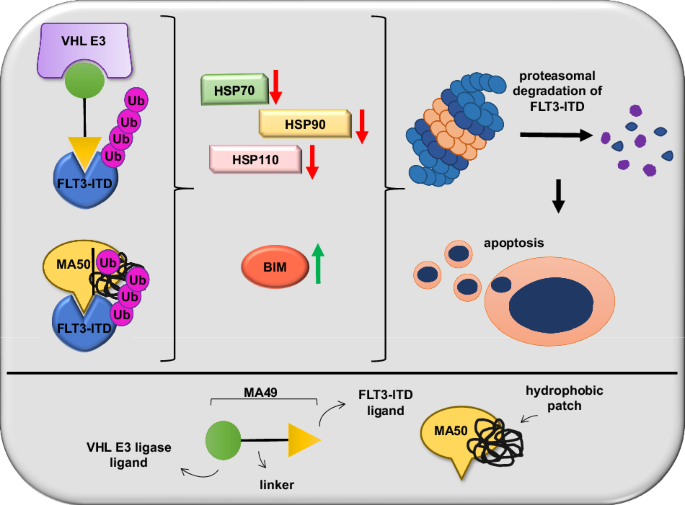
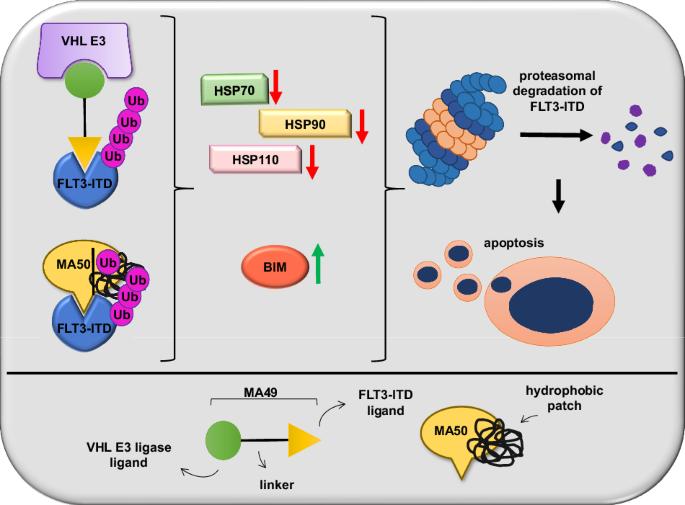
突变型FMS样酪氨酸激酶-3的选择性降解需要热休克蛋白的BIM依赖性消耗
FMS样酪氨酸激酶-3(FLT3-ITD)的内部串联重复是急性髓性白血病(AML)中常见的突变。蛋白水解靶向嵌合体(PROTACs)能诱导蛋白酶体降解突变的FLT3,是一种创新的药理学方法。除了泛素-蛋白酶体系统外,控制靶向蛋白酶解的分子机制尚未明确,而PROTACs是目前已知的唯一一种FLT3降解剂。我们报告说,基于von-Hippel-Lindau泛素连接酶的FLT3 PROTAC MA49(美洛替尼-49)和FLT3疏水标签分子MA50(哈洛替尼-50)可减少内质网相关的致癌FLT3-ITD,但不影响FLT3。纳摩尔剂量的 MA49 和 MA50 能诱导带有 FLT3-ITD 的人类白血病细胞系和原发性急性髓细胞白血病细胞凋亡(p < 0.05-0.0001),但不能诱导原发性造血干细胞和分化的免疫细胞、FLT3 野生型细胞、视网膜细胞和 c-KIT 依赖性细胞凋亡。MA49 对 FLT3-ITD 阳性白血病细胞的体内活性已在 Danio rerio 模型中得到验证。降解剂诱导的 FLT3-ITD 损失涉及促凋亡的 BH3 唯一蛋白 BIM 和先前未确定的降解剂诱导的蛋白质折叠伴侣的消耗。HSP90 和 HSP110 的表达水平与急性髓细胞性白血病患者存活率的降低相关(p < 0.1),HSP90、HSP110 和 BIM 与原发性急性髓细胞性白血病细胞中 FLT3 的表达相关(p < 0.01)。HSP90抑制了降解剂诱导的FLT3-ITD消除,从而建立了一个机理明确的反馈回路。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Leukemia
医学-血液学
CiteScore
18.10
自引率
3.50%
发文量
270
审稿时长
3-6 weeks
期刊介绍:
Title: Leukemia
Journal Overview:
Publishes high-quality, peer-reviewed research
Covers all aspects of research and treatment of leukemia and allied diseases
Includes studies of normal hemopoiesis due to comparative relevance
Topics of Interest:
Oncogenes
Growth factors
Stem cells
Leukemia genomics
Cell cycle
Signal transduction
Molecular targets for therapy
And more
Content Types:
Original research articles
Reviews
Letters
Correspondence
Comments elaborating on significant advances and covering topical issues
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


