{"title":"Photochromic reaction pathways of 1,1′-azobis-1,2,3-triazole: A CASSCF and spin-flip DFT study","authors":"Liangyue Cheng","doi":"10.1016/j.comptc.2024.114870","DOIUrl":null,"url":null,"abstract":"<div><p>This study employed multiconfigurational CASSCF and MS-CASPT2 methods to investigate the photoinduced isomerization mechanism of 1,1′-azobis-1,2,3-triazole. The MS-CASPT2//CASSCF computational results indicate that the photoisomerization reaction of 1,1′-azobis-1,2,3-triazole involves a non-adiabatic transition pathway through the S<sub>2</sub> state. After photoexcitation to the S<sub>2</sub> state, the molecule undergoes internal conversion to reach S<sub>1</sub>-min, followed by a non-radiative transition back to the ground state. The S<sub>1</sub>/S<sub>0</sub>-CI and S<sub>1</sub>-min have similar structures and close energies, which facilitates unidirectional rotation. The weak coupling between the ground state and excited states may be due to the strong electron-withdrawing nature of the N<sub>8</sub> chain. Additionally, using the MS-CASPT2//CASSCF computational results as a reference, we compared the differences in describing the photoisomerization process of this compound using the SF-DFT method, both qualitatively and quantitatively. The results demonstrate that the SF-DFT method significantly underestimates the energy of the S<sub>1</sub> state. These findings are expected to deepen the understanding of non-adiabatic transitions in the photoinduced rotation of high-nitrogen compounds and provide theoretical insights for other high-nitrogen compounds that may exhibit photochromism.</p></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1241 ","pages":"Article 114870"},"PeriodicalIF":3.0000,"publicationDate":"2024-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004092","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
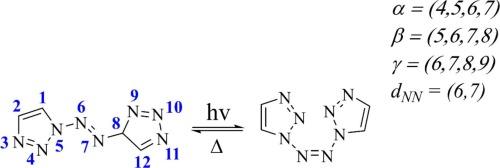
This study employed multiconfigurational CASSCF and MS-CASPT2 methods to investigate the photoinduced isomerization mechanism of 1,1′-azobis-1,2,3-triazole. The MS-CASPT2//CASSCF computational results indicate that the photoisomerization reaction of 1,1′-azobis-1,2,3-triazole involves a non-adiabatic transition pathway through the S2 state. After photoexcitation to the S2 state, the molecule undergoes internal conversion to reach S1-min, followed by a non-radiative transition back to the ground state. The S1/S0-CI and S1-min have similar structures and close energies, which facilitates unidirectional rotation. The weak coupling between the ground state and excited states may be due to the strong electron-withdrawing nature of the N8 chain. Additionally, using the MS-CASPT2//CASSCF computational results as a reference, we compared the differences in describing the photoisomerization process of this compound using the SF-DFT method, both qualitatively and quantitatively. The results demonstrate that the SF-DFT method significantly underestimates the energy of the S1 state. These findings are expected to deepen the understanding of non-adiabatic transitions in the photoinduced rotation of high-nitrogen compounds and provide theoretical insights for other high-nitrogen compounds that may exhibit photochromism.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


