The role of linkers and frustrated lewis pairs catalysts in the formation of zwitterionic 1,2-anti-addition product with non-conjugated terminal diacetylenes: A computational study
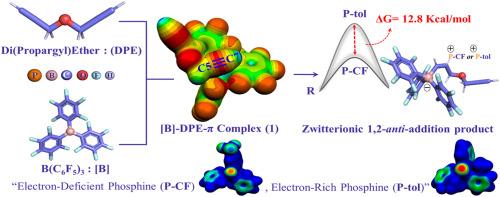
{"title":"The role of linkers and frustrated lewis pairs catalysts in the formation of zwitterionic 1,2-anti-addition product with non-conjugated terminal diacetylenes: A computational study","authors":"Tulsi R. Patel , Bishwajit Ganguly","doi":"10.1016/j.jmgm.2024.108866","DOIUrl":null,"url":null,"abstract":"<div><p>This study presents a computational investigation into the mechanistic pathway and the linker units involved in forming the zwitterionic 1,2-<em>anti</em>-addition product of non-conjugated diacetylenes, di(propargyl)ether (<strong>DPE</strong>), di(prop-2yn-1yl)sulfane (<strong>DPS</strong>) and 1,6-Heptadiyne (<strong>HD</strong>) catalyzed by the inter-molecular phosphine/borane frustrated Lewis pairs (FLPs), i.e., PPh<sub>2</sub>[C<sub>6</sub>H<sub>3</sub>(CF<sub>3</sub>)<sub>2</sub>](<strong>P-CF</strong>)/[B(C<sub>6</sub>F<sub>5</sub>)<sub>3</sub>](<strong>[B]</strong>) and P(o-tolyl)<sub>3</sub>(<strong>P-tol</strong>)/[B(C<sub>6</sub>F<sub>5</sub>)<sub>3</sub>](<strong>[B]</strong>). The potential energy surface (PES) calculations reveal that the <em>anti</em>-addition of <strong>P-CF</strong> to the internal C-atoms of acetylene units is energetically more favored than that of the addition of <strong>P-tol</strong> in <strong>DPE</strong>, <strong>DPS</strong>, and <strong>HD</strong> by ∼10.0, ∼9.2, and ∼6.0 kcal/mol, respectively. The calculations performed with <strong>DPE</strong> contain “—O—,” linker unit exhibits superior reactivity than <strong>DPS</strong> and <strong>HD</strong>, which suggests the electronegativity of linkers plays a significant role and facilitates the addition of Lewis bases. The higher electronegativity of linker units enables the 1,2-addition reaction by lowering the free energy activation barriers, as observed in the DFT calculations. The Molecular Electrostatic Potential (MESP) study shows that the electrostatic interactions favor the addition of <strong>P-CF</strong> to the active acetylene positions (<strong>C5</strong>/<strong>C4</strong>/<strong>C4</strong>) of <strong>[B]</strong>-<strong>DPE/DPS/HD</strong>-π complexes than the <strong>P-tol</strong>. The Distortion/Interaction (D/I) analysis reveals that transition states involving <strong>P-CF</strong> (TS1, TS3, and TS5) exhibit more interaction energy (ΔE<sub>Int</sub>) and less distortion energies (ΔE<sup>d</sup>) than that of the <strong>P-tol</strong> (TS2, TS4, and TS6). Further, the Energy Decomposition Analysis (EDA) also rationalizes the preferential approach of the electron-deficient Lewis base over the electron-rich one on the basis of the significant contribution of orbital interaction energies (ΔE<sub>orbital</sub>) in the cases of <strong>P-CF</strong>; TS1, TS3, and TS5. This study suggests that the electronic effects of substrates and the FLPs are crucial to facilitate the desired products formed with non-conjugated terminal alkynes.</p></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"133 ","pages":"Article 108866"},"PeriodicalIF":2.7000,"publicationDate":"2024-09-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001669","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
This study presents a computational investigation into the mechanistic pathway and the linker units involved in forming the zwitterionic 1,2-anti-addition product of non-conjugated diacetylenes, di(propargyl)ether (DPE), di(prop-2yn-1yl)sulfane (DPS) and 1,6-Heptadiyne (HD) catalyzed by the inter-molecular phosphine/borane frustrated Lewis pairs (FLPs), i.e., PPh2[C6H3(CF3)2](P-CF)/[B(C6F5)3]([B]) and P(o-tolyl)3(P-tol)/[B(C6F5)3]([B]). The potential energy surface (PES) calculations reveal that the anti-addition of P-CF to the internal C-atoms of acetylene units is energetically more favored than that of the addition of P-tol in DPE, DPS, and HD by ∼10.0, ∼9.2, and ∼6.0 kcal/mol, respectively. The calculations performed with DPE contain “—O—,” linker unit exhibits superior reactivity than DPS and HD, which suggests the electronegativity of linkers plays a significant role and facilitates the addition of Lewis bases. The higher electronegativity of linker units enables the 1,2-addition reaction by lowering the free energy activation barriers, as observed in the DFT calculations. The Molecular Electrostatic Potential (MESP) study shows that the electrostatic interactions favor the addition of P-CF to the active acetylene positions (C5/C4/C4) of [B]-DPE/DPS/HD-π complexes than the P-tol. The Distortion/Interaction (D/I) analysis reveals that transition states involving P-CF (TS1, TS3, and TS5) exhibit more interaction energy (ΔEInt) and less distortion energies (ΔEd) than that of the P-tol (TS2, TS4, and TS6). Further, the Energy Decomposition Analysis (EDA) also rationalizes the preferential approach of the electron-deficient Lewis base over the electron-rich one on the basis of the significant contribution of orbital interaction energies (ΔEorbital) in the cases of P-CF; TS1, TS3, and TS5. This study suggests that the electronic effects of substrates and the FLPs are crucial to facilitate the desired products formed with non-conjugated terminal alkynes.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


