{"title":"Are we ready for disease modification in myeloproliferative neoplasms?","authors":"Claire N. Harrison","doi":"10.1002/hem3.70003","DOIUrl":null,"url":null,"abstract":"<p>Myeloproliferative neoplasms (MPN) are chronic myeloid diseases characterized by clinical heterogeneity and disordered JAK/STAT signaling. For the purposes of this perspective, the three common MPN, essential thrombocythemia (ET), polycythemia vera (PV), and myelofibrosis (MF), will be considered.<span><sup>1</sup></span> These are closely related conditions as recognized by William Dameshek in 1951; in recent decades, multiple scientific and clinical advances have improved our understanding of their pathophysiology and outcomes for patients (<i>vida infra</i>). Risks for these patients include reduced life expectancy due to thrombosis, hemorrhage, and disease transformation (Figure 1). However, while for other myeloid malignancies, notably chronic and acute myeloid leukemia, disease modification is largely measured by morphological and molecular responses correlating with survival benefits. Currently, the situation for MPN management is perceived to be less advanced. This may in part be due to the relatively late designation of MPN as a malignancy.</p><p>Here, we shall define disease modification as treatments or interventions that affect the underlying pathophysiology of the disease and have beneficial outcomes on the clinical course and address the question: “Are we ready for disease modification in MPN?”</p><p>Major clinical events for patients with MPN as stated above are thrombosis, hemorrhage, and disease transformation; particularly to myelofibrosis, these require long-term studies to assess. But in recent years, at least for treating myelofibrosis, attention has also been focused on splenomegaly and the impact of disease-related symptoms on quality of life as relevant endpoints.</p><p>Specifically, for ET and PV treatment, algorithms focus on risk stratification based on perceived thrombotic and, to a lesser extent, hemorrhagic risk but not on reducing disease transformation. Therapeutic approaches focus on addressing modifiable factors from the perspective of vascular disease; for example, smoking, obesity, and hypertension; venesection or phlebotomy in PV; followed by antiplatelet drugs and then, in selected patients, cytoreductive therapy.<span><sup>2</sup></span> The aim of cytoreductive therapy is thus to reduce thrombosis or hemorrhage. In PV, these approaches have been shown to reduce the risk of thrombosis. For example, in the CYTOPV study, hematocrit (HCT) target <45% or 45%–50% at a median follow-up of 31 months; thrombotic events or deaths from vascular causes: <45% HCT (2.7%); 45%–50% HCT (9.8%).<span><sup>3</sup></span> Second, in the ECLAP study where low-dose aspirin compared to placebo was shown to reduce the risk of the end point of nonfatal myocardial infarction, nonfatal stroke, pulmonary embolism, major venous thrombosis, or death from cardiovascular causes: (relative risk, 0.40; 95% confidence interval, 0.18–0.91; <i>p</i> = 0.03).<span><sup>4</sup></span></p><p>In MF, the advent of JAK inhibitor-based therapy focussed treatment for the majority of patients ineligible for stem cell transplant upon reducing spleen size and symptoms. Both of these facets of MF do probably reflect underlying pathophysiology and, furthermore, spleen size reduction has been shown to correlate with overall survival advantage.<span><sup>5, 6</sup></span> Interestingly, momelotinib, a JAK1, JAK2, and ACVR inhibitor, treated patients who were independent of transfusions and also had a survival benefit.<span><sup>7</sup></span> Arguably, survival benefit reflects disease modification but here duration of benefit for spleen and anemia are limited. Also, there is no robust evidence that these agents substantially modify underlying disease if this was defined as clonal response, fibrosis resolution, and so forth.<span><sup>8</sup></span> In the context of MF therapy, the drive to improve patient outcomes has resulted in combination trials. Unfortunately, two recent phase 3 clinical trials, one with the BET inhibitor pelabresib and the other with the BCL<sub>XL</sub> inhibitor navitoclax, have been judged to have failed based on a lack of delivering an incremental benefit for symptom improvement as a mandated end-point.<span><sup>9, 10</sup></span> Further data from these studies will hopefully show survival benefits linked to more biological endpoints related to spleen, clonality, fibrosis, and other responses. These data should hopefully influence a paradigm shift for the regulatory agencies and the field toward a focus instead of disease modification but this will certainly require data extending beyond the recent standard of 24 weeks.</p><p>This will be discussed in the context of PV. The oft-used term “low-risk” PV is misleading; since these patients do have risks greater than the normal population. For example, a large multicenter retrospective study of low-risk PV patients reported that the annual post diagnosis thrombosis rate was 2.62%.<span><sup>11</sup></span> Publications reporting standardized mortality rates suggest that young low-risk PV patients have substantial longer-term disease-related risks (thrombosis as well as disease transformation) which were also supported by a meta-analysis.<span><sup>12</sup></span> This paper also concluded that newer therapies, in particular, newer formulations of interferon alpha, which are for the most part less toxic should strongly be considered. This proposal is supported by two recent cohort analyses. In the first cohort where PV patients were treated with interferon α had superior both myelofibrosis-free and overall survival. Here, in low-risk patients, 20-year myelofibrosis-free survival was 66%, and for interferon α, hydroxycarbamide and no cytoreduction was 84%, 65%, and 55% respectively (<i>p</i> < 0.001); 20-year overall survival was 87% for the whole cohort, and 100%, 85%, and 80%, respectively, for interferon alpha, hydroxycarbamide, and no cytoreduction (<i>p</i> = 0.44).<span><sup>13</sup></span> In the second cohort of ET and PV patients diagnosed before the age of 25 years, myelofibrosis-free survival was 100% and superior for interferon α-treated patients at a median follow-up of 9 years.<span><sup>14</sup></span></p><p>In the second line setting, the MAJIC PV study evaluated the long-term benefits of ruxolitinib (JAK1 and JAK2 inhibitor) in 180 patients who were resistant or intolerant to hydroxycarbamide (a very high risk population).<span><sup>15</sup></span> Here, event-free survival (EFS, defined as major thrombosis, hemorrhage, transformation, and death) was superior for patients attaining complete response within 1 year (HR, 0.41; 95% CI, 0.21–0.78; <i>p</i> = 0.01); and those on ruxolitinib (HR, 0.58; 95% CI, 0.35–0.94; <i>p</i> = 0.03). Perhaps, more importantly, serial analysis of <i>JAK2V617F</i> variant allele fraction revealed molecular response (50% reduction) was more frequent with ruxolitinib and was associated with improved outcomes (progression-free survival [PFS] <i>p</i> = 0.001, EFS <i>p</i> = 0.001, overall survival <i>p</i> = 0.01) and clearance of <i>JAK2V617F</i> stem/progenitor cells. For the MPN field, this is the first time in a prospective study that molecular response to a therapy (albeit a modest one) has been linked to survival outcomes.</p><p>In PV, a recent analysis of the PROUD PV, a study (mainly high-risk PV) which treated patients with rho-pegylated interferon alfa-2b suggested that the latter was superior to control therapies in producing event-free survival including thrombosis, hemorrhage, transformation, and death.<span><sup>16</sup></span> At the European Hematology Association (EHA) 2024 meeting, this benefit was also linked to molecular response.<span><sup>17</sup></span></p><p>In conclusion, these data for PV patients suggest that disease modification could be defined by a molecular response (at least a 50% reduction in <i>JAK2</i>V617F variant allele frequency which is achievable with interferon α and also ruxolitinib. Furthermore, a re-assessment of the benefit of treating the majority of low-risk PV with these drugs with lower toxicity should be considered.<span><sup>18</sup></span></p><p>In MF, a more modest molecular response, a 20% reduction in driver (specifics not available) mutation (variant allele frequency, VAF) in patients treated with either imetelstat, a telomerase inhibitor, or navitoclax was linked to overall survival.<span><sup>19, 20</sup></span> The situation with other driver mutations is unclear; for example, in the MAJIC ET study,<span><sup>21</sup></span> one patient lost their <i>CALR</i> mutation and yet progressed. There are no available data for patients with <i>MPL</i> mutations.</p><p>The use of molecular response as a marker of disease modification requires significant focus on standardization and confirmation of the required thresholds and how this can be translated into patient benefit. We have much to learn from other disease areas in particular chronic myeloid leukemia (CML) and acute myeloid leukemia (AML). In the MPN context, different clonal architectures and dynamics may also be important. Intriguing recent data suggest that patients with MPN may harbor driver mutations in particular <i>JAK2V617F</i> from an early age perhaps having the mutation for several decades. This suggests that if widespread genetic screening is introduced, even more young patients with these mutations will be identified.<span><sup>22, 23</sup></span> The existence of clones for many years and questions about the residual normal stem cell pool raise the question that perhaps a minimal disease state may be acceptable versus eradication at all costs. Referencing again data for PV colleagues from France have suggested that hematological control and reducing the <i>JAK2V617F</i> VAF to less than 10% can lead to a prolonged treatment-free remission.<span><sup>24</sup></span> However, if an ultimate molecular endpoint was to drive the clone to a minimal state, data suggest that those with a <i>JAK2</i> clonal hematopoiesis of indeterminate potential (CHIP) have a high risk of cardiovascular disease<span><sup>25</sup></span> needs to be considered. Less data are available regarding the risks of very low-level <i>CALR</i> and <i>MPL</i> clones both of which are less frequently identified as CHIP mutations.</p><p>Having explored the concept that disease modification in PV and perhaps MF might be linked to molecular response for ET, the situation is less clear. Perhaps, ultimately, <i>JAK2-</i>ET could be managed in a similar way to PV and indeed prognostic scores such as ISPET identify <i>JAK2</i>-ET as associated with more thrombotic risk than <i>CALR</i>-ET though the latter has more risk of MF-transformation. In the future, we might treat <i>CALR-</i>, <i>MPL</i>- and <i>JAK2-</i>ET differently. Triple-negative ET has a different much more benign disease trajectory and our future strategy may be not to treat this group of patients.<span><sup>26</sup></span> Thus, for now, our likely focus will remain on PV and MF.</p><p>Finally, concerning molecular response, it is important to also consider clonal complexity, the presence of more than one pathogenic mutation which may or may not occur in the same clone as the driver mutation. This represents another aspect of the work which needs to be done in assessing this endpoint and the impact of therapies.</p><p>In addition to spleen, symptoms, and molecular response, other potential markers of disease modification in MF include changes in cytokines, changes in the inflammatory milieu, and assessment of the bone marrow histology using AI-based techniques. Each of these will require significant evaluation and standardization. Considering spleen response, the time to loss of response may be important as initial spleen volume reduction appears robustly linked to survival benefits. For symptoms, we should not lose the benefits we have already delivered for our patients but would propose that the need to recognize a non-inferiority endpoint may be more appropriate.</p><p>As discussed above, recent studies in MF have cast further doubt upon current therapeutic targets such as spleen and symptom response as trial endpoints. Newer agents may more directly address the underlying pathophysiology of the disease. Specifically, these currently include mutant CALR-directed therapies such as vaccination, mut-CALR targeted antibodies, bispecific agents, and perhaps also CAR-T therapies and mutation-specific JAK2 inhibitors. These therapies offer exciting new opportunities to truly modify disease but we will need to advance our understanding of how this benefit can be measured. Interestingly, these therapies may also reinvigorate the debate concerning the classification of MPN subtypes toward a more mutation-specific approach as supported by the work of Grinfeld and colleagues.<span><sup>27</sup></span> Here, in an MPN cohort comprising mainly ET patients, Grinfeld and colleagues collated 63 clinical and genomic variables to create prognostic models and generated individual predictions of patient outcomes which may also be used to guide treatment. Importantly, they demonstrated that the outcome for patients was very much driven by the genomic variable. Therefore, a further implication of this model is that we might abandon the clinical descriptively defined MPN entities (ET, PV, MF) from the late 19th and early 20th century. Also, this model offers the tantalizing opportunity to use such data sets to generate a real-world evidence-based “digital twin” of a patient and their predicted outcome for a clinical trial.</p><p>Disease modification for MPN should no longer be considered an elusive goal; however, it is a complex one. Careful evidence-based current management can achieve this, for example, attaining a complete response (control of blood parameters and spleen size) can lead to event-free survival. But in the future, in PV, we may move toward treating almost all patients, especially with more modern less toxic therapies (such as ruxolitinib or rog-pegylated interferon-α-2b), with the aim of achieving a 50% reduction in <i>JAK2</i> VAF or molecular response once nuances of standardization, optimal kinetics of response, and so forth, have been addressed. Meanwhile, we need to gather further data concerning the benefits or not of molecular response for <i>CALR</i> and <i>MPL</i>-mutated diseases.</p><p>Carefully collated and annotated clinical information coupled with the evolution of newer therapies will hopefully drive the development of robust data to generate better endpoints in the context of the more heterogeneous disorders ET and MF as well as the less well-defined MPN entities such as pre-fibrotic myelofibrosis.</p><p>Meanwhile, there is much work to be done which will require close collaboration between patient communities, clinical experts, translation scientists, pharmaceutical companies, approval agencies, and payers as well as those skilled in interrogating massive data sets.</p><p>This article was solely written by the author with no additional support. The figure was produced with support from HemaSphere.</p><p>Claire N. Harrison: Research funding: Celgene (BMS), Constellation, GSK, Novartis. Advisory role: AbbVie, AOP, BMS, CTI, IMAGO, Incyte, Novartis, Galacteo, Geron, GSK, Incyte, Janssen, Keros, MSD, SOBI, Morphosys, and is an Editor of HemaSphere.</p><p>This research received no funding.</p>","PeriodicalId":12982,"journal":{"name":"HemaSphere","volume":"8 9","pages":""},"PeriodicalIF":7.6000,"publicationDate":"2024-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/hem3.70003","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"HemaSphere","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/hem3.70003","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
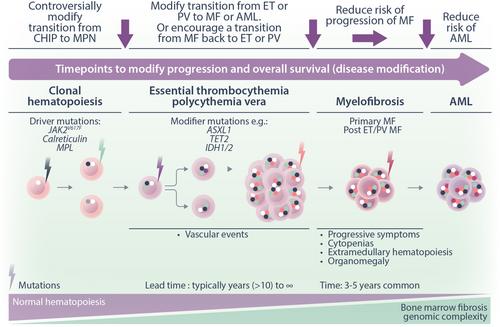
Myeloproliferative neoplasms (MPN) are chronic myeloid diseases characterized by clinical heterogeneity and disordered JAK/STAT signaling. For the purposes of this perspective, the three common MPN, essential thrombocythemia (ET), polycythemia vera (PV), and myelofibrosis (MF), will be considered.1 These are closely related conditions as recognized by William Dameshek in 1951; in recent decades, multiple scientific and clinical advances have improved our understanding of their pathophysiology and outcomes for patients (vida infra). Risks for these patients include reduced life expectancy due to thrombosis, hemorrhage, and disease transformation (Figure 1). However, while for other myeloid malignancies, notably chronic and acute myeloid leukemia, disease modification is largely measured by morphological and molecular responses correlating with survival benefits. Currently, the situation for MPN management is perceived to be less advanced. This may in part be due to the relatively late designation of MPN as a malignancy.
Here, we shall define disease modification as treatments or interventions that affect the underlying pathophysiology of the disease and have beneficial outcomes on the clinical course and address the question: “Are we ready for disease modification in MPN?”
Major clinical events for patients with MPN as stated above are thrombosis, hemorrhage, and disease transformation; particularly to myelofibrosis, these require long-term studies to assess. But in recent years, at least for treating myelofibrosis, attention has also been focused on splenomegaly and the impact of disease-related symptoms on quality of life as relevant endpoints.
Specifically, for ET and PV treatment, algorithms focus on risk stratification based on perceived thrombotic and, to a lesser extent, hemorrhagic risk but not on reducing disease transformation. Therapeutic approaches focus on addressing modifiable factors from the perspective of vascular disease; for example, smoking, obesity, and hypertension; venesection or phlebotomy in PV; followed by antiplatelet drugs and then, in selected patients, cytoreductive therapy.2 The aim of cytoreductive therapy is thus to reduce thrombosis or hemorrhage. In PV, these approaches have been shown to reduce the risk of thrombosis. For example, in the CYTOPV study, hematocrit (HCT) target <45% or 45%–50% at a median follow-up of 31 months; thrombotic events or deaths from vascular causes: <45% HCT (2.7%); 45%–50% HCT (9.8%).3 Second, in the ECLAP study where low-dose aspirin compared to placebo was shown to reduce the risk of the end point of nonfatal myocardial infarction, nonfatal stroke, pulmonary embolism, major venous thrombosis, or death from cardiovascular causes: (relative risk, 0.40; 95% confidence interval, 0.18–0.91; p = 0.03).4
In MF, the advent of JAK inhibitor-based therapy focussed treatment for the majority of patients ineligible for stem cell transplant upon reducing spleen size and symptoms. Both of these facets of MF do probably reflect underlying pathophysiology and, furthermore, spleen size reduction has been shown to correlate with overall survival advantage.5, 6 Interestingly, momelotinib, a JAK1, JAK2, and ACVR inhibitor, treated patients who were independent of transfusions and also had a survival benefit.7 Arguably, survival benefit reflects disease modification but here duration of benefit for spleen and anemia are limited. Also, there is no robust evidence that these agents substantially modify underlying disease if this was defined as clonal response, fibrosis resolution, and so forth.8 In the context of MF therapy, the drive to improve patient outcomes has resulted in combination trials. Unfortunately, two recent phase 3 clinical trials, one with the BET inhibitor pelabresib and the other with the BCLXL inhibitor navitoclax, have been judged to have failed based on a lack of delivering an incremental benefit for symptom improvement as a mandated end-point.9, 10 Further data from these studies will hopefully show survival benefits linked to more biological endpoints related to spleen, clonality, fibrosis, and other responses. These data should hopefully influence a paradigm shift for the regulatory agencies and the field toward a focus instead of disease modification but this will certainly require data extending beyond the recent standard of 24 weeks.
This will be discussed in the context of PV. The oft-used term “low-risk” PV is misleading; since these patients do have risks greater than the normal population. For example, a large multicenter retrospective study of low-risk PV patients reported that the annual post diagnosis thrombosis rate was 2.62%.11 Publications reporting standardized mortality rates suggest that young low-risk PV patients have substantial longer-term disease-related risks (thrombosis as well as disease transformation) which were also supported by a meta-analysis.12 This paper also concluded that newer therapies, in particular, newer formulations of interferon alpha, which are for the most part less toxic should strongly be considered. This proposal is supported by two recent cohort analyses. In the first cohort where PV patients were treated with interferon α had superior both myelofibrosis-free and overall survival. Here, in low-risk patients, 20-year myelofibrosis-free survival was 66%, and for interferon α, hydroxycarbamide and no cytoreduction was 84%, 65%, and 55% respectively (p < 0.001); 20-year overall survival was 87% for the whole cohort, and 100%, 85%, and 80%, respectively, for interferon alpha, hydroxycarbamide, and no cytoreduction (p = 0.44).13 In the second cohort of ET and PV patients diagnosed before the age of 25 years, myelofibrosis-free survival was 100% and superior for interferon α-treated patients at a median follow-up of 9 years.14
In the second line setting, the MAJIC PV study evaluated the long-term benefits of ruxolitinib (JAK1 and JAK2 inhibitor) in 180 patients who were resistant or intolerant to hydroxycarbamide (a very high risk population).15 Here, event-free survival (EFS, defined as major thrombosis, hemorrhage, transformation, and death) was superior for patients attaining complete response within 1 year (HR, 0.41; 95% CI, 0.21–0.78; p = 0.01); and those on ruxolitinib (HR, 0.58; 95% CI, 0.35–0.94; p = 0.03). Perhaps, more importantly, serial analysis of JAK2V617F variant allele fraction revealed molecular response (50% reduction) was more frequent with ruxolitinib and was associated with improved outcomes (progression-free survival [PFS] p = 0.001, EFS p = 0.001, overall survival p = 0.01) and clearance of JAK2V617F stem/progenitor cells. For the MPN field, this is the first time in a prospective study that molecular response to a therapy (albeit a modest one) has been linked to survival outcomes.
In PV, a recent analysis of the PROUD PV, a study (mainly high-risk PV) which treated patients with rho-pegylated interferon alfa-2b suggested that the latter was superior to control therapies in producing event-free survival including thrombosis, hemorrhage, transformation, and death.16 At the European Hematology Association (EHA) 2024 meeting, this benefit was also linked to molecular response.17
In conclusion, these data for PV patients suggest that disease modification could be defined by a molecular response (at least a 50% reduction in JAK2V617F variant allele frequency which is achievable with interferon α and also ruxolitinib. Furthermore, a re-assessment of the benefit of treating the majority of low-risk PV with these drugs with lower toxicity should be considered.18
In MF, a more modest molecular response, a 20% reduction in driver (specifics not available) mutation (variant allele frequency, VAF) in patients treated with either imetelstat, a telomerase inhibitor, or navitoclax was linked to overall survival.19, 20 The situation with other driver mutations is unclear; for example, in the MAJIC ET study,21 one patient lost their CALR mutation and yet progressed. There are no available data for patients with MPL mutations.
The use of molecular response as a marker of disease modification requires significant focus on standardization and confirmation of the required thresholds and how this can be translated into patient benefit. We have much to learn from other disease areas in particular chronic myeloid leukemia (CML) and acute myeloid leukemia (AML). In the MPN context, different clonal architectures and dynamics may also be important. Intriguing recent data suggest that patients with MPN may harbor driver mutations in particular JAK2V617F from an early age perhaps having the mutation for several decades. This suggests that if widespread genetic screening is introduced, even more young patients with these mutations will be identified.22, 23 The existence of clones for many years and questions about the residual normal stem cell pool raise the question that perhaps a minimal disease state may be acceptable versus eradication at all costs. Referencing again data for PV colleagues from France have suggested that hematological control and reducing the JAK2V617F VAF to less than 10% can lead to a prolonged treatment-free remission.24 However, if an ultimate molecular endpoint was to drive the clone to a minimal state, data suggest that those with a JAK2 clonal hematopoiesis of indeterminate potential (CHIP) have a high risk of cardiovascular disease25 needs to be considered. Less data are available regarding the risks of very low-level CALR and MPL clones both of which are less frequently identified as CHIP mutations.
Having explored the concept that disease modification in PV and perhaps MF might be linked to molecular response for ET, the situation is less clear. Perhaps, ultimately, JAK2-ET could be managed in a similar way to PV and indeed prognostic scores such as ISPET identify JAK2-ET as associated with more thrombotic risk than CALR-ET though the latter has more risk of MF-transformation. In the future, we might treat CALR-, MPL- and JAK2-ET differently. Triple-negative ET has a different much more benign disease trajectory and our future strategy may be not to treat this group of patients.26 Thus, for now, our likely focus will remain on PV and MF.
Finally, concerning molecular response, it is important to also consider clonal complexity, the presence of more than one pathogenic mutation which may or may not occur in the same clone as the driver mutation. This represents another aspect of the work which needs to be done in assessing this endpoint and the impact of therapies.
In addition to spleen, symptoms, and molecular response, other potential markers of disease modification in MF include changes in cytokines, changes in the inflammatory milieu, and assessment of the bone marrow histology using AI-based techniques. Each of these will require significant evaluation and standardization. Considering spleen response, the time to loss of response may be important as initial spleen volume reduction appears robustly linked to survival benefits. For symptoms, we should not lose the benefits we have already delivered for our patients but would propose that the need to recognize a non-inferiority endpoint may be more appropriate.
As discussed above, recent studies in MF have cast further doubt upon current therapeutic targets such as spleen and symptom response as trial endpoints. Newer agents may more directly address the underlying pathophysiology of the disease. Specifically, these currently include mutant CALR-directed therapies such as vaccination, mut-CALR targeted antibodies, bispecific agents, and perhaps also CAR-T therapies and mutation-specific JAK2 inhibitors. These therapies offer exciting new opportunities to truly modify disease but we will need to advance our understanding of how this benefit can be measured. Interestingly, these therapies may also reinvigorate the debate concerning the classification of MPN subtypes toward a more mutation-specific approach as supported by the work of Grinfeld and colleagues.27 Here, in an MPN cohort comprising mainly ET patients, Grinfeld and colleagues collated 63 clinical and genomic variables to create prognostic models and generated individual predictions of patient outcomes which may also be used to guide treatment. Importantly, they demonstrated that the outcome for patients was very much driven by the genomic variable. Therefore, a further implication of this model is that we might abandon the clinical descriptively defined MPN entities (ET, PV, MF) from the late 19th and early 20th century. Also, this model offers the tantalizing opportunity to use such data sets to generate a real-world evidence-based “digital twin” of a patient and their predicted outcome for a clinical trial.
Disease modification for MPN should no longer be considered an elusive goal; however, it is a complex one. Careful evidence-based current management can achieve this, for example, attaining a complete response (control of blood parameters and spleen size) can lead to event-free survival. But in the future, in PV, we may move toward treating almost all patients, especially with more modern less toxic therapies (such as ruxolitinib or rog-pegylated interferon-α-2b), with the aim of achieving a 50% reduction in JAK2 VAF or molecular response once nuances of standardization, optimal kinetics of response, and so forth, have been addressed. Meanwhile, we need to gather further data concerning the benefits or not of molecular response for CALR and MPL-mutated diseases.
Carefully collated and annotated clinical information coupled with the evolution of newer therapies will hopefully drive the development of robust data to generate better endpoints in the context of the more heterogeneous disorders ET and MF as well as the less well-defined MPN entities such as pre-fibrotic myelofibrosis.
Meanwhile, there is much work to be done which will require close collaboration between patient communities, clinical experts, translation scientists, pharmaceutical companies, approval agencies, and payers as well as those skilled in interrogating massive data sets.
This article was solely written by the author with no additional support. The figure was produced with support from HemaSphere.
Claire N. Harrison: Research funding: Celgene (BMS), Constellation, GSK, Novartis. Advisory role: AbbVie, AOP, BMS, CTI, IMAGO, Incyte, Novartis, Galacteo, Geron, GSK, Incyte, Janssen, Keros, MSD, SOBI, Morphosys, and is an Editor of HemaSphere.
期刊介绍:
HemaSphere, as a publication, is dedicated to disseminating the outcomes of profoundly pertinent basic, translational, and clinical research endeavors within the field of hematology. The journal actively seeks robust studies that unveil novel discoveries with significant ramifications for hematology.
In addition to original research, HemaSphere features review articles and guideline articles that furnish lucid synopses and discussions of emerging developments, along with recommendations for patient care.
Positioned as the foremost resource in hematology, HemaSphere augments its offerings with specialized sections like HemaTopics and HemaPolicy. These segments engender insightful dialogues covering a spectrum of hematology-related topics, including digestible summaries of pivotal articles, updates on new therapies, deliberations on European policy matters, and other noteworthy news items within the field. Steering the course of HemaSphere are Editor in Chief Jan Cools and Deputy Editor in Chief Claire Harrison, alongside the guidance of an esteemed Editorial Board comprising international luminaries in both research and clinical realms, each representing diverse areas of hematologic expertise.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


