Predicting Peptide Ionization Efficiencies for Electrospray Ionization Mass Spectrometry Using Machine Learning
IF 3.1
2区 化学
Q2 BIOCHEMICAL RESEARCH METHODS
Journal of the American Society for Mass Spectrometry
Pub Date : 2024-09-09
DOI:10.1021/jasms.4c00137
引用次数: 0
Abstract
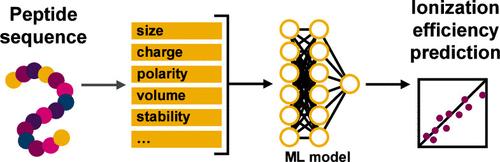
Mass spectrometry (MS) is inherently an information-rich technique. In this era of big data, label-free MS quantification for nontargeted studies has gained increasing popularity, especially for complex systems. One of the cornerstones of successful label-free quantification is the predictive modeling of ionization efficiency (IE) based on solutes’ physicochemical properties. While many have studied IE modeling for small molecules, there are limited reports on peptide IEs. In this study, we leverage the stoichiometric relationship in trypsin digests of well-characterized monoclonal antibodies (mAbs) to compile a data set of relative ionization efficiencies (RIEs) for 241 peptides. From each peptide’s sequence, we computed a set of physiochemical descriptors, which were then used to train machine learning regression models to predict RIEs. Peptides shorter than 20 amino acids had RIEs that were highly correlated to their molecular weight. A random forest (RF) model was able to best predict the RIEs of a test data set with a mean relative error of 23.9%. For larger peptides, a multilayer perceptron (MLP) model improved RIE prediction compared to current best practices, reducing mean relative error from 60.5% to 32.0%. Finally, we also show the application of the RF model in label-free relative protein quantification and improving the quantification of peptide post-translational modifications (PTMs). This approach to predicting peptide IEs from their sequences enables the development of accurate label-free quantification workflows for peptide and protein analysis.
利用机器学习预测电喷雾离子化质谱仪的肽离子化效率
质谱(MS)本身就是一种信息丰富的技术。在这个大数据时代,用于非靶向研究的无标记质谱定量方法越来越受欢迎,尤其是在复杂系统中。成功实现无标记定量的基石之一是根据溶质的物理化学特性对电离效率(IE)进行预测建模。虽然许多人研究了小分子的离子化效率建模,但有关多肽离子化效率的报道却很有限。在这项研究中,我们利用特性良好的单克隆抗体(mAbs)的胰蛋白酶消化物中的化学计量关系,为 241 种肽编制了相对电离效率(RIE)数据集。根据每种肽的序列,我们计算出了一组理化描述因子,然后利用这些描述因子训练机器学习回归模型来预测相对电离效率。短于 20 个氨基酸的多肽的 RIE 与其分子量高度相关。随机森林(RF)模型能够最好地预测测试数据集的 RIE,平均相对误差为 23.9%。对于较大的多肽,多层感知器(MLP)模型与当前的最佳实践相比改善了 RIE 预测,将平均相对误差从 60.5% 降至 32.0%。最后,我们还展示了 RF 模型在无标记蛋白质相对定量中的应用,并改进了肽翻译后修饰 (PTM) 的定量。这种从序列预测多肽 IEs 的方法有助于为多肽和蛋白质分析开发精确的无标记定量工作流程。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
5.50
自引率
9.40%
发文量
257
审稿时长
1 months
期刊介绍:
The Journal of the American Society for Mass Spectrometry presents research papers covering all aspects of mass spectrometry, incorporating coverage of fields of scientific inquiry in which mass spectrometry can play a role.
Comprehensive in scope, the journal publishes papers on both fundamentals and applications of mass spectrometry. Fundamental subjects include instrumentation principles, design, and demonstration, structures and chemical properties of gas-phase ions, studies of thermodynamic properties, ion spectroscopy, chemical kinetics, mechanisms of ionization, theories of ion fragmentation, cluster ions, and potential energy surfaces. In addition to full papers, the journal offers Communications, Application Notes, and Accounts and Perspectives
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


