{"title":"Nature of the chemical bonding and electronic structure of dicoordinated copper(I) complexes of alkenes, alkynes, and NHC ligands: a DFT overview","authors":"Nadjet Aimene, Abdallah Zaiter, Hacene Nemdili, Bachir Zouchoune","doi":"10.1007/s11224-024-02366-6","DOIUrl":null,"url":null,"abstract":"<div><p>A DFT (density functional theory) investigation using the generalized gradient approximation BP86 and the hybrid B3LYP functionals and TZP basis set is dealing with the bonding, the electronic structure and the interaction types occurred within the XCuL’ and (LCuL’)<sup>+</sup> (X = Cl, CH<sub>3</sub>, CN, CF<sub>3</sub>, L = CO, NH<sub>3</sub>, PH<sub>3</sub>, and L’ = C<sub>2</sub>H<sub>2</sub>, C<sub>2</sub>H<sub>4</sub>, C<sub>4</sub>H<sub>6</sub>, C<sub>6</sub>H<sub>6</sub>, and NHC) complexes. The optimized structures and energy decomposition analysis of XCuL’ and (LCuL’)<sup>+</sup> complexes were employed to provide a relationship between the bond lengths, the X-Cu-L’ and L-Cu-L’ bond angles, the Wiberg indices, the Mayer bond orders, interaction energies, and the Cu-L’ bonding character. The energy decomposition analysis indicates that the interactions occurred for various L’ ligands are more electrostatically than covalently bonded to the Cu(I) center formally of + I oxidation state. The different contributions stemming from electrostatic and orbital interactions are significant, in relationship with the ionic and covalent characters, respectively. The contribution from σ-donation to the bonding energy was found more important for the NHC ligand than the alkene and alkynes ones. However, the contribution from π-back-donation was found to be comparable for all complexes. The σ-bonding contributes more than 50% into the total orbital interaction overtaking those of π type, in accordance with the population of the copper 4s orbital, particularly in the presence of C<sub>6</sub>H<sub>6</sub> and NHC ligands. The interactions in all complexes exhibit comparable deformation densities and NOCV orbital shapes. Besides, it has been shown that the ΔE<sub>prep</sub> contributes weakly in the deformation of the interacting fragments as well as the BSSE correction which impacts weakly or negligibly the interactions between the fragments composing different XCuL’ and (LCuL’)<sup>+</sup> complexes.</p></div>","PeriodicalId":780,"journal":{"name":"Structural Chemistry","volume":"36 1","pages":"269 - 284"},"PeriodicalIF":2.1000,"publicationDate":"2024-08-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structural Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s11224-024-02366-6","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
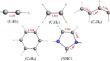
A DFT (density functional theory) investigation using the generalized gradient approximation BP86 and the hybrid B3LYP functionals and TZP basis set is dealing with the bonding, the electronic structure and the interaction types occurred within the XCuL’ and (LCuL’)+ (X = Cl, CH3, CN, CF3, L = CO, NH3, PH3, and L’ = C2H2, C2H4, C4H6, C6H6, and NHC) complexes. The optimized structures and energy decomposition analysis of XCuL’ and (LCuL’)+ complexes were employed to provide a relationship between the bond lengths, the X-Cu-L’ and L-Cu-L’ bond angles, the Wiberg indices, the Mayer bond orders, interaction energies, and the Cu-L’ bonding character. The energy decomposition analysis indicates that the interactions occurred for various L’ ligands are more electrostatically than covalently bonded to the Cu(I) center formally of + I oxidation state. The different contributions stemming from electrostatic and orbital interactions are significant, in relationship with the ionic and covalent characters, respectively. The contribution from σ-donation to the bonding energy was found more important for the NHC ligand than the alkene and alkynes ones. However, the contribution from π-back-donation was found to be comparable for all complexes. The σ-bonding contributes more than 50% into the total orbital interaction overtaking those of π type, in accordance with the population of the copper 4s orbital, particularly in the presence of C6H6 and NHC ligands. The interactions in all complexes exhibit comparable deformation densities and NOCV orbital shapes. Besides, it has been shown that the ΔEprep contributes weakly in the deformation of the interacting fragments as well as the BSSE correction which impacts weakly or negligibly the interactions between the fragments composing different XCuL’ and (LCuL’)+ complexes.
期刊介绍:
Structural Chemistry is an international forum for the publication of peer-reviewed original research papers that cover the condensed and gaseous states of matter and involve numerous techniques for the determination of structure and energetics, their results, and the conclusions derived from these studies. The journal overcomes the unnatural separation in the current literature among the areas of structure determination, energetics, and applications, as well as builds a bridge to other chemical disciplines. Ist comprehensive coverage encompasses broad discussion of results, observation of relationships among various properties, and the description and application of structure and energy information in all domains of chemistry.
We welcome the broadest range of accounts of research in structural chemistry involving the discussion of methodologies and structures,experimental, theoretical, and computational, and their combinations. We encourage discussions of structural information collected for their chemicaland biological significance.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


