Qinghe Gao, Tammo Dukker, Artur M. Schweidtmann and Jana M. Weber
{"title":"Self-supervised graph neural networks for polymer property prediction†","authors":"Qinghe Gao, Tammo Dukker, Artur M. Schweidtmann and Jana M. Weber","doi":"10.1039/D4ME00088A","DOIUrl":null,"url":null,"abstract":"<p >The estimation of polymer properties is of crucial importance in many domains such as energy, healthcare, and packaging. Recently, graph neural networks (GNNs) have shown promising results for the prediction of polymer properties based on supervised learning. However, the training of GNNs in a supervised learning task demands a huge amount of polymer property data that is time-consuming and computationally/experimentally expensive to obtain. Self-supervised learning offers great potential to reduce this data demand through pre-training the GNNs on polymer structure data only. These pre-trained GNNs can then be fine-tuned on the supervised property prediction task using a much smaller labeled dataset. We propose to leverage self-supervised learning techniques in GNNs for the prediction of polymer properties. We employ a recent polymer graph representation that includes essential features of polymers, such as monomer combinations, stochastic chain architecture, and monomer stoichiometry, and process the polymer graphs through a tailored GNN architecture. We investigate three self-supervised learning setups: (i) node- and edge-level pre-training, (ii) graph-level pre-training, and (iii) ensembled node-, edge- & graph-level pre-training. We additionally explore three different transfer strategies of fully connected layers with the GNN architecture. Our results indicate that the ensemble node-, edge- & graph-level self-supervised learning with all layers transferred depicts the best performance across dataset size. In scarce data scenarios, it decreases the root mean square errors by 28.39% and 19.09% for the prediction of electron affinity and ionization potential compared to supervised learning without the pre-training task.</p>","PeriodicalId":91,"journal":{"name":"Molecular Systems Design & Engineering","volume":" 11","pages":" 1130-1143"},"PeriodicalIF":3.2000,"publicationDate":"2024-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/me/d4me00088a?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Design & Engineering","FirstCategoryId":"5","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/me/d4me00088a","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
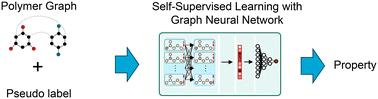
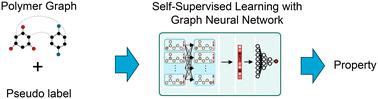
The estimation of polymer properties is of crucial importance in many domains such as energy, healthcare, and packaging. Recently, graph neural networks (GNNs) have shown promising results for the prediction of polymer properties based on supervised learning. However, the training of GNNs in a supervised learning task demands a huge amount of polymer property data that is time-consuming and computationally/experimentally expensive to obtain. Self-supervised learning offers great potential to reduce this data demand through pre-training the GNNs on polymer structure data only. These pre-trained GNNs can then be fine-tuned on the supervised property prediction task using a much smaller labeled dataset. We propose to leverage self-supervised learning techniques in GNNs for the prediction of polymer properties. We employ a recent polymer graph representation that includes essential features of polymers, such as monomer combinations, stochastic chain architecture, and monomer stoichiometry, and process the polymer graphs through a tailored GNN architecture. We investigate three self-supervised learning setups: (i) node- and edge-level pre-training, (ii) graph-level pre-training, and (iii) ensembled node-, edge- & graph-level pre-training. We additionally explore three different transfer strategies of fully connected layers with the GNN architecture. Our results indicate that the ensemble node-, edge- & graph-level self-supervised learning with all layers transferred depicts the best performance across dataset size. In scarce data scenarios, it decreases the root mean square errors by 28.39% and 19.09% for the prediction of electron affinity and ionization potential compared to supervised learning without the pre-training task.
期刊介绍:
Molecular Systems Design & Engineering provides a hub for cutting-edge research into how understanding of molecular properties, behaviour and interactions can be used to design and assemble better materials, systems, and processes to achieve specific functions. These may have applications of technological significance and help address global challenges.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


