Michael Tynes, Michael G. Taylor, Jan Janssen, Daniel J. Burrill, Danny Perez, Ping Yang and Nicholas Lubbers
{"title":"Linear graphlet models for accurate and interpretable cheminformatics†","authors":"Michael Tynes, Michael G. Taylor, Jan Janssen, Daniel J. Burrill, Danny Perez, Ping Yang and Nicholas Lubbers","doi":"10.1039/D4DD00089G","DOIUrl":null,"url":null,"abstract":"<p >Advances in machine learning have given rise to a plurality of data-driven methods for predicting chemical properties from molecular structure. For many decades, the cheminformatics field has relied heavily on structural fingerprinting, while in recent years much focus has shifted toward leveraging highly parameterized deep neural networks which usually maximize accuracy. Beyond accuracy, to be useful and trustworthy in scientific applications, machine learning techniques often need intuitive explanations for model predictions and uncertainty quantification techniques so a practitioner might know when a model is appropriate to apply to new data. Here we revisit graphlet histogram fingerprints and introduce several new elements. We show that linear models built on graphlet fingerprints attain accuracy that is competitive with the state of the art while retaining an explainability advantage over black-box approaches. We show how to produce precise explanations of predictions by exploiting the relationships between molecular graphlets and show that these explanations are consistent with chemical intuition, experimental measurements, and theoretical calculations. Finally, we show how to use the presence of unseen fragments in new molecules to adjust predictions and quantify uncertainty.</p>","PeriodicalId":72816,"journal":{"name":"Digital discovery","volume":" 10","pages":" 1980-1996"},"PeriodicalIF":6.2000,"publicationDate":"2024-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/dd/d4dd00089g?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Digital discovery","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/dd/d4dd00089g","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
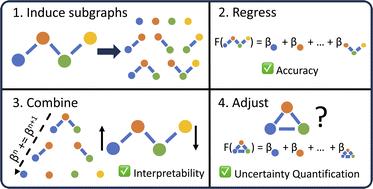
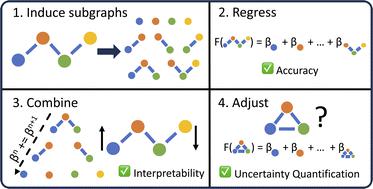
Advances in machine learning have given rise to a plurality of data-driven methods for predicting chemical properties from molecular structure. For many decades, the cheminformatics field has relied heavily on structural fingerprinting, while in recent years much focus has shifted toward leveraging highly parameterized deep neural networks which usually maximize accuracy. Beyond accuracy, to be useful and trustworthy in scientific applications, machine learning techniques often need intuitive explanations for model predictions and uncertainty quantification techniques so a practitioner might know when a model is appropriate to apply to new data. Here we revisit graphlet histogram fingerprints and introduce several new elements. We show that linear models built on graphlet fingerprints attain accuracy that is competitive with the state of the art while retaining an explainability advantage over black-box approaches. We show how to produce precise explanations of predictions by exploiting the relationships between molecular graphlets and show that these explanations are consistent with chemical intuition, experimental measurements, and theoretical calculations. Finally, we show how to use the presence of unseen fragments in new molecules to adjust predictions and quantify uncertainty.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


