{"title":"Co-expression network and survival analysis of breast cancer inflammation and immune system hallmark genes","authors":"Ayaka Yakushi , Masahiro Sugimoto , Takanori Sasaki","doi":"10.1016/j.compbiolchem.2024.108204","DOIUrl":null,"url":null,"abstract":"<div><p>The tertiary lymphoid structure (TLS) plays a central role in cancer immune response, and its gene expression pattern, called the TLS signature, has shown prognostic value in breast cancer. The formation of TLS and tumor-associated high endothelial venules (TA-HEVs), responsible for lymphocytic infiltration within the TLS, is associated with the expression of cancer hallmark genes (CHGs) related to immunity and inflammation. In this study, we performed co-expression network analysis of immune- and inflammation-related CHGs to identify predictive genes for breast cancer. In total, 382 immune- and inflammation-related CHGs with high expression variance were extracted from the GSE86166 microarray dataset of patients with breast cancer. CHGs were classified into five modules by applying weighted gene co-expression network analysis. The survival analysis results for each module showed that one module comprising 45 genes was statistically significant for relapse-free and overall survival. Four network properties identified key genes in this module with high prognostic prediction abilities: <em>CD34</em>, <em>CXCL12</em>, <em>F2RL2</em>, <em>JAM2</em>, <em>PROS1</em>, <em>RAPGEF3</em>, and <em>SELP</em>. The prognostic accuracy of the seven genes in breast cancer was synergistic and exceeded that of other predictors in both small and large public datasets. Enrichment analysis predicted that these genes had functions related to leukocyte infiltration of TA-HEVs. There was a positive correlation between key gene expression and the TLS signature, suggesting that gene expression levels are associated with TLS density. Co-expression network analysis of inflammation- and immune-related CHGs allowed us to identify genes that share a standard function in cancer immunity and have a high prognostic predictive value. This analytical approach may contribute to the identification of prognostic genes in TLS.</p></div>","PeriodicalId":10616,"journal":{"name":"Computational Biology and Chemistry","volume":"113 ","pages":"Article 108204"},"PeriodicalIF":2.6000,"publicationDate":"2024-09-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S1476927124001920/pdfft?md5=1c407a366ff621563e7bacfdd48a6bb7&pid=1-s2.0-S1476927124001920-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Biology and Chemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1476927124001920","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 0
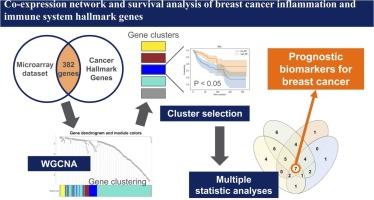
Abstract
The tertiary lymphoid structure (TLS) plays a central role in cancer immune response, and its gene expression pattern, called the TLS signature, has shown prognostic value in breast cancer. The formation of TLS and tumor-associated high endothelial venules (TA-HEVs), responsible for lymphocytic infiltration within the TLS, is associated with the expression of cancer hallmark genes (CHGs) related to immunity and inflammation. In this study, we performed co-expression network analysis of immune- and inflammation-related CHGs to identify predictive genes for breast cancer. In total, 382 immune- and inflammation-related CHGs with high expression variance were extracted from the GSE86166 microarray dataset of patients with breast cancer. CHGs were classified into five modules by applying weighted gene co-expression network analysis. The survival analysis results for each module showed that one module comprising 45 genes was statistically significant for relapse-free and overall survival. Four network properties identified key genes in this module with high prognostic prediction abilities: CD34, CXCL12, F2RL2, JAM2, PROS1, RAPGEF3, and SELP. The prognostic accuracy of the seven genes in breast cancer was synergistic and exceeded that of other predictors in both small and large public datasets. Enrichment analysis predicted that these genes had functions related to leukocyte infiltration of TA-HEVs. There was a positive correlation between key gene expression and the TLS signature, suggesting that gene expression levels are associated with TLS density. Co-expression network analysis of inflammation- and immune-related CHGs allowed us to identify genes that share a standard function in cancer immunity and have a high prognostic predictive value. This analytical approach may contribute to the identification of prognostic genes in TLS.
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


