John S. Wang, Rebecca L. Koch, Daniel Kenney-Jung, Erin Huggins, Sirajbir S. Sodhi, Andrew P. Landstrom, Dilraj S. Grewal, Priya S. Kishnani
{"title":"Gaucher disease type 3c: Expanding the clinical spectrum of an ultra-rare disease","authors":"John S. Wang, Rebecca L. Koch, Daniel Kenney-Jung, Erin Huggins, Sirajbir S. Sodhi, Andrew P. Landstrom, Dilraj S. Grewal, Priya S. Kishnani","doi":"10.1002/jmd2.12440","DOIUrl":null,"url":null,"abstract":"<p>Gaucher disease (GD) type 3 is an autosomal recessive lysosomal disease caused by deficiency of β-glucocerebrosidase (GCase) and encompasses a spectrum of cardiac, neurological, and ophthalmological abnormalities. Although the clinical presentations can be diverse, a recognized clinical trajectory points to an early onset, predominantly before 18 years. GD type 3c is primarily caused by homozygosity for <i>GBA</i> pathogenic variant c.1342G>C (p.Asp448His; historically referred to as D409H) and includes visceral, hematological, skeletal, and cardiac abnormalities. Notably, GD type 3c is distinct from other GD types because it is primarily characterized by valvular heart disease. Yet, with less than 50 patients with GD type 3c reported to date, the phenotypic spectrum and extent of cardiac involvement remains ill-defined. We present a 20-year-old female with an atypical presentation of GD type 3c consisting of chronic intermediate uveitis as the presenting feature and the presence of extensive polyneuropathy starting in adolescence which has been previously unreported in GD type 3c. Distinctively, she has maintained normal cardiac function. Moreover, we compare our case with those reported in the literature to broaden awareness of the varied initial presentations of this disease. The diverse presentations seen in GD type 3c, underscored by our case and those previously reported, demonstrate the need for standardized evaluation and management protocols.</p>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"65 5","pages":"313-322"},"PeriodicalIF":1.8000,"publicationDate":"2024-08-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12440","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12440","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
Abstract
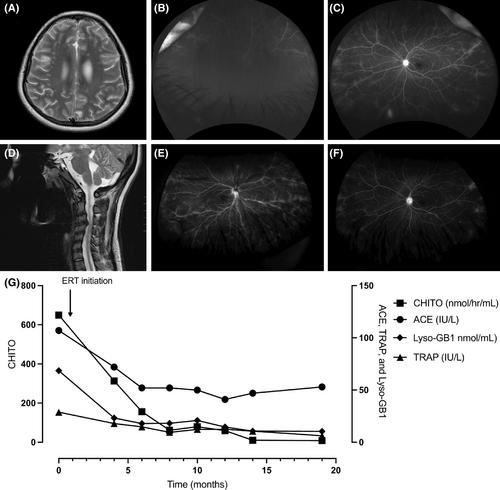
Gaucher disease (GD) type 3 is an autosomal recessive lysosomal disease caused by deficiency of β-glucocerebrosidase (GCase) and encompasses a spectrum of cardiac, neurological, and ophthalmological abnormalities. Although the clinical presentations can be diverse, a recognized clinical trajectory points to an early onset, predominantly before 18 years. GD type 3c is primarily caused by homozygosity for GBA pathogenic variant c.1342G>C (p.Asp448His; historically referred to as D409H) and includes visceral, hematological, skeletal, and cardiac abnormalities. Notably, GD type 3c is distinct from other GD types because it is primarily characterized by valvular heart disease. Yet, with less than 50 patients with GD type 3c reported to date, the phenotypic spectrum and extent of cardiac involvement remains ill-defined. We present a 20-year-old female with an atypical presentation of GD type 3c consisting of chronic intermediate uveitis as the presenting feature and the presence of extensive polyneuropathy starting in adolescence which has been previously unreported in GD type 3c. Distinctively, she has maintained normal cardiac function. Moreover, we compare our case with those reported in the literature to broaden awareness of the varied initial presentations of this disease. The diverse presentations seen in GD type 3c, underscored by our case and those previously reported, demonstrate the need for standardized evaluation and management protocols.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


