{"title":"Element-specific descriptors to predict the stability of binary nanoalloys","authors":"","doi":"10.1016/j.commatsci.2024.113336","DOIUrl":null,"url":null,"abstract":"<div><p>The practical applications of nanoalloys, which are known for their exceptional catalytic activity, are difficult to realize owing to their intricate stability of these systems, which is influenced by structural variations, configurational nuances, and elemental interactions. Many combinations resulting from the inclusion of many different possible constituent elements intensifies the complexity of their analysis, emphasizing the need for accurate stability prediction methods. This study investigated the stability of A−B binary nanoalloys composed of 3<em>d</em>, 4<em>d</em>, and 5<em>d</em> late transition metal elements such as Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, and Au. Density functional theory (DFT) calculations and supervised learning (SL) were employed to predict the stability of these alloys. The excess energy, an indicator used to evaluate the stability of nanoalloys, was predicted using the structure- and element-specific descriptors in a two-stage SL method. The first SL stage involves expressing the excess energy through structure-specific descriptors such as bond fractions and element deviation within each coordination number (CN). The second SL stage involves expressing the regression coefficients of the structure-specific descriptors using element-specific descriptors. The element-specific descriptors predicting the element deviation in each CN correspond to differences in melting point and atomic radius. Simultaneously, the prediction of bond fractions relies on factors such as electronegativity difference and electron density discontinuity between the constituent elements. The study findings suggest that the stability of a nanoalloy can be broadly categorized into that of its surface and inner components. Monte Carlo simulations based on structure- and element-specific descriptors exhibit the capability to predict the stable configurations of binary nanoalloys without the need for DFT. The approach described in this study significantly enhances the efficiency with which these calculations may be executed, thereby expediting the analysis of the properties of these alloys.</p></div>","PeriodicalId":10650,"journal":{"name":"Computational Materials Science","volume":null,"pages":null},"PeriodicalIF":3.1000,"publicationDate":"2024-09-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Materials Science","FirstCategoryId":"88","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0927025624005573","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
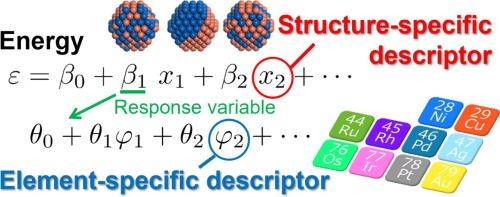
Abstract
The practical applications of nanoalloys, which are known for their exceptional catalytic activity, are difficult to realize owing to their intricate stability of these systems, which is influenced by structural variations, configurational nuances, and elemental interactions. Many combinations resulting from the inclusion of many different possible constituent elements intensifies the complexity of their analysis, emphasizing the need for accurate stability prediction methods. This study investigated the stability of A−B binary nanoalloys composed of 3d, 4d, and 5d late transition metal elements such as Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt, and Au. Density functional theory (DFT) calculations and supervised learning (SL) were employed to predict the stability of these alloys. The excess energy, an indicator used to evaluate the stability of nanoalloys, was predicted using the structure- and element-specific descriptors in a two-stage SL method. The first SL stage involves expressing the excess energy through structure-specific descriptors such as bond fractions and element deviation within each coordination number (CN). The second SL stage involves expressing the regression coefficients of the structure-specific descriptors using element-specific descriptors. The element-specific descriptors predicting the element deviation in each CN correspond to differences in melting point and atomic radius. Simultaneously, the prediction of bond fractions relies on factors such as electronegativity difference and electron density discontinuity between the constituent elements. The study findings suggest that the stability of a nanoalloy can be broadly categorized into that of its surface and inner components. Monte Carlo simulations based on structure- and element-specific descriptors exhibit the capability to predict the stable configurations of binary nanoalloys without the need for DFT. The approach described in this study significantly enhances the efficiency with which these calculations may be executed, thereby expediting the analysis of the properties of these alloys.
期刊介绍:
The goal of Computational Materials Science is to report on results that provide new or unique insights into, or significantly expand our understanding of, the properties of materials or phenomena associated with their design, synthesis, processing, characterization, and utilization. To be relevant to the journal, the results should be applied or applicable to specific material systems that are discussed within the submission.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


