Loss of DOCK2 potentiates Inflammatory Bowel Disease–associated colorectal cancer via immune dysfunction and IFNγ induction of IDO1 expression
IF 6.9
1区 医学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
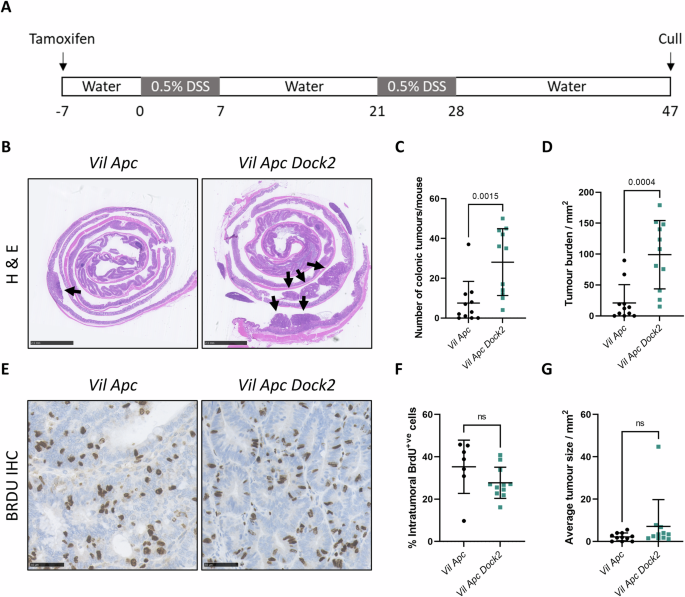
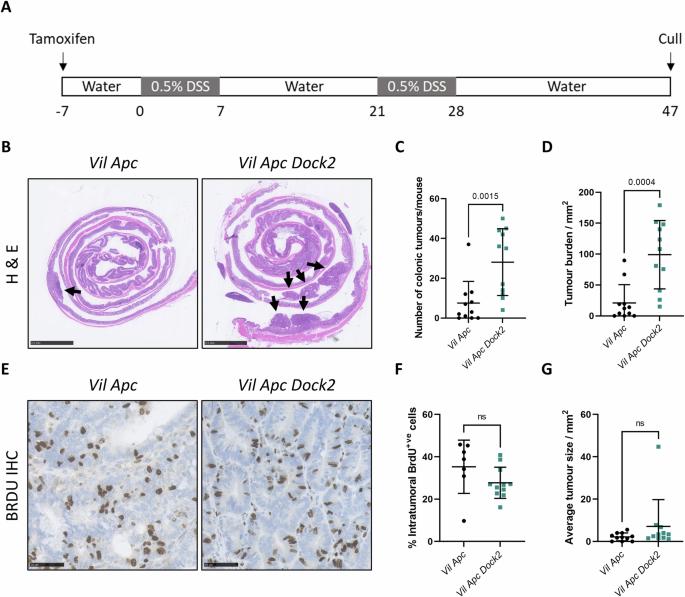
Inflammatory Bowel Disease-associated colorectal cancer (IBD-CRC) is a known and serious complication of Inflammatory Bowel Disease (IBD) affecting the colon. However, relatively little is known about the pathogenesis of IBD-associated colorectal cancer in comparison with its sporadic cancer counterpart. Here, we investigated the function of Dock2, a gene mutated in ~10% of IBD-associated colorectal cancers that encodes a guanine nucleotide exchange factor (GEF). Using a genetically engineered mouse model of IBD-CRC, we found that whole body loss of Dock2 increases tumourigenesis via immune dysregulation. Dock2-deficient tumours displayed increased levels of IFNγ-associated genes, including the tryptophan metabolising, immune modulatory enzyme, IDO1, when compared to Dock2-proficient tumours. This phenotype was driven by increased IFNγ-production in T cell populations, which infiltrated Dock2-deficient tumours, promoting IDO1 expression in tumour epithelial cells. We show that IDO1 inhibition delays tumourigenesis in Dock2 knockout mice, and we confirm that this pathway is conserved across species as IDO1 expression is elevated in human IBD-CRC and in sporadic CRC cases with mutated DOCK2. Together, these data demonstrate a previously unidentified tumour suppressive role of DOCK2 that limits IFNγ-induced IDO1 expression and cancer progression, opening potential new avenues for therapeutic intervention.
DOCK2 的缺失会通过免疫功能紊乱和 IFNγ 诱导 IDO1 的表达,增强炎症性肠病相关性结直肠癌的发病率。
众所周知,炎症性肠病相关性结直肠癌(IBD-CRC)是炎症性肠病(IBD)影响结肠的一种严重并发症。然而,与散发性结直肠癌相比,人们对 IBD 相关性结直肠癌的发病机制知之甚少。在这里,我们研究了 Dock2 的功能。Dock2 是一种编码鸟嘌呤核苷酸交换因子(GEF)的基因,在约 10% 的 IBD 相关性结直肠癌中发生突变。通过使用基因工程小鼠 IBD-CRC 模型,我们发现 Dock2 的全身缺失会通过免疫失调增加肿瘤发生。与Dock2缺陷型肿瘤相比,Dock2缺陷型肿瘤的IFNγ相关基因水平升高,包括色氨酸代谢免疫调节酶IDO1。这种表型是由T细胞群中IFNγ的分泌增加驱动的,T细胞群渗入Dock2缺陷型肿瘤,促进了IDO1在肿瘤上皮细胞中的表达。我们的研究表明,抑制 IDO1 可延缓 Dock2 基因敲除小鼠的肿瘤发生,我们还证实了这一通路在不同物种中是保守的,因为在人类 IBD-CRC 和有突变 DOCK2 的散发性 CRC 病例中,IDO1 的表达都升高了。这些数据共同证明了 DOCK2 先前未被发现的肿瘤抑制作用,它限制了 IFNγ 诱导的 IDO1 表达和癌症进展,为治疗干预开辟了潜在的新途径。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Oncogene
医学-生化与分子生物学
CiteScore
15.30
自引率
1.20%
发文量
404
审稿时长
1 months
期刊介绍:
Oncogene is dedicated to advancing our understanding of cancer processes through the publication of exceptional research. The journal seeks to disseminate work that challenges conventional theories and contributes to establishing new paradigms in the etio-pathogenesis, diagnosis, treatment, or prevention of cancers. Emphasis is placed on research shedding light on processes driving metastatic spread and providing crucial insights into cancer biology beyond existing knowledge.
Areas covered include the cellular and molecular biology of cancer, resistance to cancer therapies, and the development of improved approaches to enhance survival. Oncogene spans the spectrum of cancer biology, from fundamental and theoretical work to translational, applied, and clinical research, including early and late Phase clinical trials, particularly those with biologic and translational endpoints.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


