CRISPR/Cas9-mediated exon skipping to restore premature translation termination in a DFNB4 mouse model
IF 4.5
3区 医学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
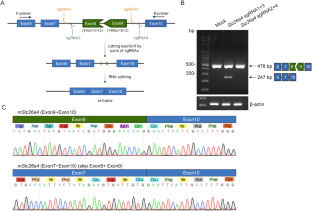
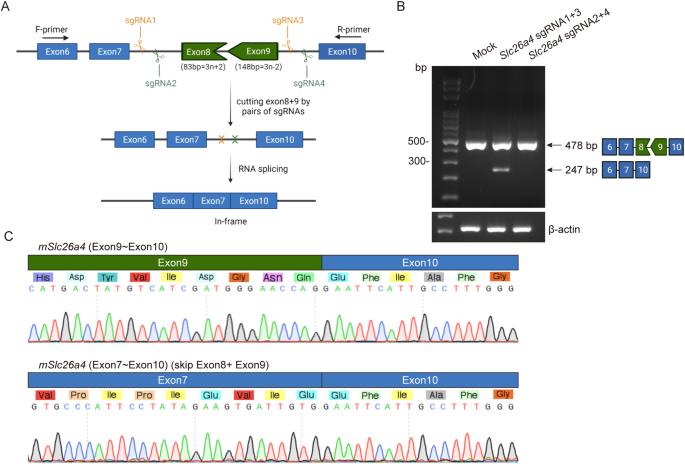
SLC26A4 encodes pendrin, a crucial anion exchanger essential for maintaining hearing function. Mutations in SLC26A4, including the prevalent c.919-2 A > G splice-site mutation among East Asian individuals, can disrupt inner ear electrolyte balance, leading to syndromic and non-syndromic hearing loss, such as Pendred syndrome and DFNB4. To explore potential therapeutic strategies, we utilized CRISPR/Cas9-mediated exon skipping to create a Slc26a4∆E8+E9/∆E8+E9 mouse model. We assessed pendrin expression in the inner ear and evaluated vestibular and auditory functions. The Slc26a4∆E8+E9/∆E8+E9 mice demonstrated reframed pendrin in the inner ear and normal vestibular functions, contrasting with severely abnormal vestibular functions observed in the Slc26a4 c.919-2 A > G splicing mutation mouse model. However, despite these molecular achievements, hearing function did not show the expected improvement, consistent with observed pathology, including cochlear hair cell loss and elevated hearing thresholds. Consequently, our findings highlight the necessity for alternative genetic editing strategies to address hearing loss caused by the SLC26A4 c.919-2 A > G mutation.
CRISPR/Cas9介导的外显子跳接可恢复 DFNB4 小鼠模型的过早翻译终止。
SLC26A4 编码pendrin,这是一种对维持听力功能至关重要的阴离子交换剂。SLC26A4 的突变,包括东亚人中普遍存在的 c.919-2 A > G 拼接位点突变,会破坏内耳电解质平衡,导致综合征和非综合征性听力损失,如 Pendred 综合征和 DFNB4。为了探索潜在的治疗策略,我们利用 CRISPR/Cas9 介导的外显子跳过技术创建了 Slc26a4∆E8+E9/∆E8+E9 小鼠模型。我们评估了pendrin在内耳中的表达,并评估了前庭和听觉功能。Slc26a4ΔE8+E9/ΔE8+E9小鼠的内耳中重构了pendrin,前庭功能正常,这与Slc26a4 c.919-2 A > G剪接突变小鼠模型中观察到的前庭功能严重异常形成了鲜明对比。然而,尽管取得了这些分子成就,听觉功能并没有出现预期的改善,这与观察到的病理变化一致,包括耳蜗毛细胞缺失和听阈升高。因此,我们的研究结果突出表明,有必要采用替代基因编辑策略来解决 SLC26A4 c.919-2 A > G 突变导致的听力损失问题。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Gene Therapy
医学-生化与分子生物学
CiteScore
9.70
自引率
2.00%
发文量
67
审稿时长
4-8 weeks
期刊介绍:
Gene Therapy covers both the research and clinical applications of novel therapeutic techniques based on a genetic component. Over the last few decades, significant advances in technologies ranging from identifying novel genetic targets that cause disease through to clinical studies, which show therapeutic benefit, have elevated this multidisciplinary field to the forefront of modern medicine.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


