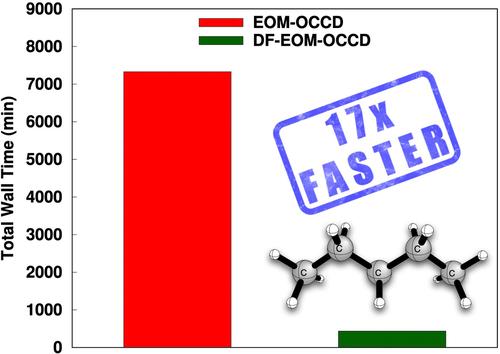
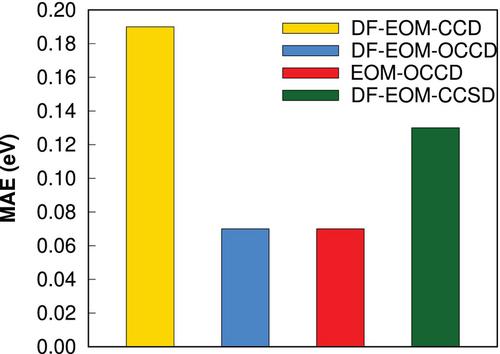
{"title":"Equation-of-motion orbital-optimized coupled-cluster doubles method with the density-fitting approximation: An efficient implementation","authors":"Aslı Ünal, Uğur Bozkaya","doi":"10.1002/jcc.27495","DOIUrl":null,"url":null,"abstract":"<p>Orbital-optimized coupled-cluster methods are very helpful for theoretical predictions of the molecular properties of challenging chemical systems, such as excited states. In this research, an efficient implementation of the equation-of-motion orbital-optimized coupled-cluster doubles method with the density-fitting (DF) approach, denoted by DF-EOM-OCCD, is presented. The computational cost of the DF-EOM-OCCD method for excitation energies is compared with that of the conventional EOM-OCCD method. Our results demonstrate that DF-EOM-OCCD excitation energies are dramatically accelerated compared to EOM-OCCD. There are almost 17-fold reductions for the <span></span><math>\n <mrow>\n <msub>\n <mrow>\n <mtext>C</mtext>\n </mrow>\n <mrow>\n <mn>5</mn>\n </mrow>\n </msub>\n <msub>\n <mrow>\n <mtext>H</mtext>\n </mrow>\n <mrow>\n <mn>12</mn>\n </mrow>\n </msub>\n </mrow></math> molecule in an aug-cc-pVTZ basis set with the RHF reference. This dramatic performance improvement comes from the reduced cost of integral transformation with the DF approach and the efficient evaluation of the particle-particle ladder (PPL) term, which is the most expensive term to evaluate. Further, our results show that the DF-EOM-OCCD approach is very helpful for the computation of excitation energies in open-shell molecular systems. Overall, we conclude that our new DF-EOM-OCCD implementation is very promising for the study of excited states in large-sized challenging chemical systems.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2969-2978"},"PeriodicalIF":3.4000,"publicationDate":"2024-09-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27495","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27495","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
Orbital-optimized coupled-cluster methods are very helpful for theoretical predictions of the molecular properties of challenging chemical systems, such as excited states. In this research, an efficient implementation of the equation-of-motion orbital-optimized coupled-cluster doubles method with the density-fitting (DF) approach, denoted by DF-EOM-OCCD, is presented. The computational cost of the DF-EOM-OCCD method for excitation energies is compared with that of the conventional EOM-OCCD method. Our results demonstrate that DF-EOM-OCCD excitation energies are dramatically accelerated compared to EOM-OCCD. There are almost 17-fold reductions for the molecule in an aug-cc-pVTZ basis set with the RHF reference. This dramatic performance improvement comes from the reduced cost of integral transformation with the DF approach and the efficient evaluation of the particle-particle ladder (PPL) term, which is the most expensive term to evaluate. Further, our results show that the DF-EOM-OCCD approach is very helpful for the computation of excitation energies in open-shell molecular systems. Overall, we conclude that our new DF-EOM-OCCD implementation is very promising for the study of excited states in large-sized challenging chemical systems.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


