Stability and Electronic Properties of K‐Sb and Na‐Sb Binary Crystals from High‐Throughput Ab Initio Calculations
IF 2.9
4区 工程技术
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
Abstract
The study of the fundamental properties of alkali antimonide photocathodes for particle accelerators is currently hindered by the limited purity of the samples. First‐principles studies can effectively complement experiments to gain insight into the stability and the electronic structure of these compounds. In this high‐throughput analysis based on density‐functional theory (DFT), two families of binary crystals with K‐Sb and Na‐Sb compositions expected to form during evaporation of multi‐alkali antimonide photocathodes are investigated. Starting from an initial pool of structures mined from existing computational databases, automatized routines included in the in‐house developed library
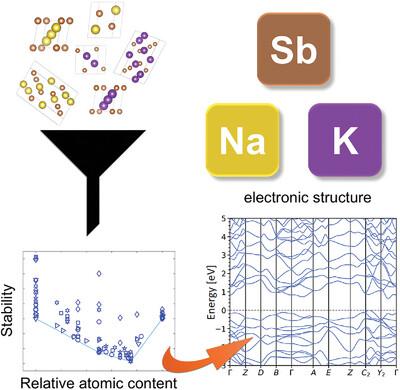
高通量 Ab Initio 计算得出的 K-Sb 和 Na-Sb 二元晶体的稳定性和电子特性
目前,用于粒子加速器的碱锑化物光电阴极的基本特性研究因样品纯度有限而受到阻碍。第一原理研究可以有效地补充实验,从而深入了解这些化合物的稳定性和电子结构。在这项基于密度泛函理论(DFT)的高通量分析中,研究了多碱锑化物光电阴极蒸发过程中预计会形成的 K-Sb 和 Na-Sb 成分二元晶体的两个系列。从现有计算数据库中挖掘的初始结构库开始,利用内部开发的程序库 aim2dat 中包含的自动例程来确定上述系统的稳定性和电子特性。通过分析形成能,对结构进行凸壳排序,保留其晶体排列信息。接着,分析了所选稳定化合物的能带结构和投影态密度。采用 r2SCAN 函数进行 DFT 计算,获得了带隙特征和大小的可靠估计值,并结合晶体中的相对碱含量进行了讨论。这些结果为预测和描述多碱锑化物光电阴极生长过程中形成的二元相提供了有用的指示。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Advanced Theory and Simulations
Multidisciplinary-Multidisciplinary
CiteScore
5.50
自引率
3.00%
发文量
221
期刊介绍:
Advanced Theory and Simulations is an interdisciplinary, international, English-language journal that publishes high-quality scientific results focusing on the development and application of theoretical methods, modeling and simulation approaches in all natural science and medicine areas, including:
materials, chemistry, condensed matter physics
engineering, energy
life science, biology, medicine
atmospheric/environmental science, climate science
planetary science, astronomy, cosmology
method development, numerical methods, statistics
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


