{"title":"Enhancing protein-ligand binding affinity prediction through sequential fusion of graph and convolutional neural networks","authors":"Yimin Yang, Ruiqin Zhang, Zijing Lin","doi":"10.1002/jcc.27499","DOIUrl":null,"url":null,"abstract":"<p>Predicting protein-ligand binding affinity is a crucial and challenging task in structure-based drug discovery. With the accumulation of complex structures and binding affinity data, various machine-learning scoring functions, particularly those based on deep learning, have been developed for this task, exhibiting superiority over their traditional counterparts. A fusion model sequentially connecting a graph neural network (GNN) and a convolutional neural network (CNN) to predict protein-ligand binding affinity is proposed in this work. In this model, the intermediate outputs of the GNN layers, as supplementary descriptors of atomic chemical environments at different levels, are concatenated with the input features of CNN. The model demonstrates a noticeable improvement in performance on CASF-2016 benchmark compared to its constituent CNN models. The generalization ability of the model is evaluated by setting a series of thresholds for ligand extended-connectivity fingerprint similarity or protein sequence similarity between the training and test sets. Masking experiment reveals that model can capture key interaction regions. Furthermore, the fusion model is applied to a virtual screening task for a novel target, PI5P4Kα. The fusion strategy significantly improves the ability of the constituent CNN model to identify active compounds. This work offers a novel approach to enhancing the accuracy of deep learning models in predicting binding affinity through fusion strategies.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2929-2940"},"PeriodicalIF":3.4000,"publicationDate":"2024-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27499","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
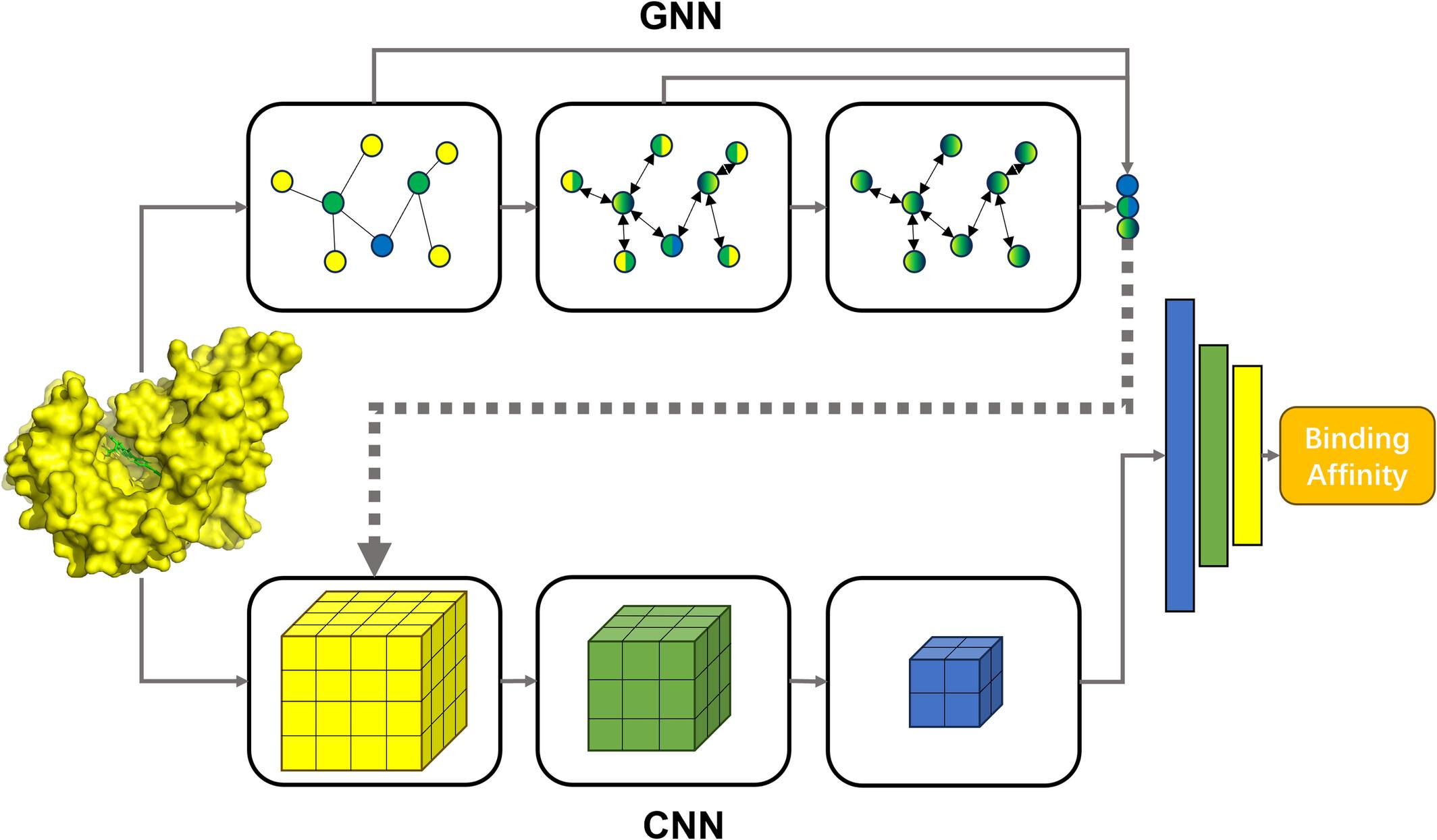
Predicting protein-ligand binding affinity is a crucial and challenging task in structure-based drug discovery. With the accumulation of complex structures and binding affinity data, various machine-learning scoring functions, particularly those based on deep learning, have been developed for this task, exhibiting superiority over their traditional counterparts. A fusion model sequentially connecting a graph neural network (GNN) and a convolutional neural network (CNN) to predict protein-ligand binding affinity is proposed in this work. In this model, the intermediate outputs of the GNN layers, as supplementary descriptors of atomic chemical environments at different levels, are concatenated with the input features of CNN. The model demonstrates a noticeable improvement in performance on CASF-2016 benchmark compared to its constituent CNN models. The generalization ability of the model is evaluated by setting a series of thresholds for ligand extended-connectivity fingerprint similarity or protein sequence similarity between the training and test sets. Masking experiment reveals that model can capture key interaction regions. Furthermore, the fusion model is applied to a virtual screening task for a novel target, PI5P4Kα. The fusion strategy significantly improves the ability of the constituent CNN model to identify active compounds. This work offers a novel approach to enhancing the accuracy of deep learning models in predicting binding affinity through fusion strategies.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


