Muzi Li, La Chen, Fangfang Yu, Huijuan Mei, Xingxing Ma, Keshuo Ding, Yanan Yang, Ziye Rong
{"title":"CTDSPL2 promotes the progression of non-small lung cancer through PI3K/AKT signaling via JAK1.","authors":"Muzi Li, La Chen, Fangfang Yu, Huijuan Mei, Xingxing Ma, Keshuo Ding, Yanan Yang, Ziye Rong","doi":"10.1038/s41420-024-02162-5","DOIUrl":null,"url":null,"abstract":"<p><p>Carboxy-terminal domain small phosphatase like 2 (CTDSPL2), one of the haloacid dehalogenase phosphatases, is associated with several diseases including cancer. However, the role of CTDSPL2 and its regulatory mechanism in lung cancer remain unclear. Here, we aimed to explore the clinical implications, biological functions, and molecular mechanisms of CTDSPL2 in non-small cell lung cancer (NSCLC). CTDSPL2 was identified as a novel target of the tumor suppressor miR-193a-3p. CTDSPL2 expression was significantly elevated in NSCLC tissues. Database analysis showed that CTDSPL2 expression was negatively correlated with patient survival. Depletion of CTDSPL2 inhibited the proliferation, migration, and invasion of NSCLC cells, as well as tumor growth and metastasis in mouse models. Additionally, silencing of CTDSPL2 enhanced CD4<sup>+</sup> T cell infiltration into tumors. Moreover, CTDSPL2 interacted with JAK1 and positively regulated JAK1 expression. Subsequent experiments indicated that CTDSPL2 activated the PI3K/AKT signaling pathway through the upregulation of JAK1, thereby promoting the progression of NSCLC. In conclusion, CTDSPL2 may play an oncogenic role in NSCLC progression by activating PI3K/AKT signaling via JAK1. These findings may provide a potential target for the diagnosis and treatment of NSCLC.</p>","PeriodicalId":9735,"journal":{"name":"Cell Death Discovery","volume":"10 1","pages":"389"},"PeriodicalIF":6.1000,"publicationDate":"2024-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11362329/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Death Discovery","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41420-024-02162-5","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
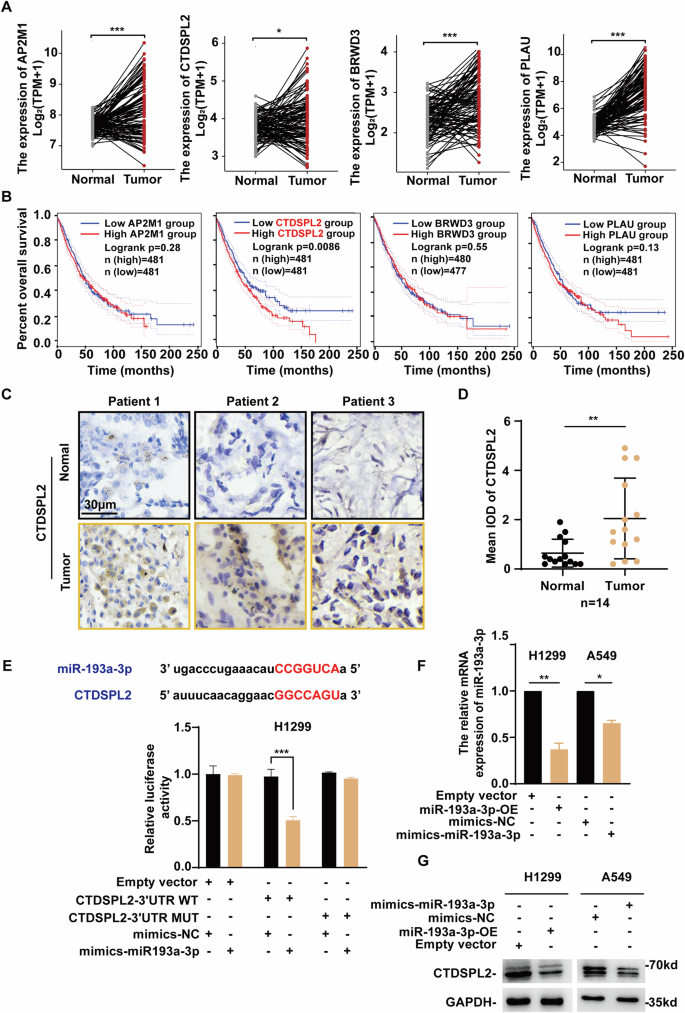
Carboxy-terminal domain small phosphatase like 2 (CTDSPL2), one of the haloacid dehalogenase phosphatases, is associated with several diseases including cancer. However, the role of CTDSPL2 and its regulatory mechanism in lung cancer remain unclear. Here, we aimed to explore the clinical implications, biological functions, and molecular mechanisms of CTDSPL2 in non-small cell lung cancer (NSCLC). CTDSPL2 was identified as a novel target of the tumor suppressor miR-193a-3p. CTDSPL2 expression was significantly elevated in NSCLC tissues. Database analysis showed that CTDSPL2 expression was negatively correlated with patient survival. Depletion of CTDSPL2 inhibited the proliferation, migration, and invasion of NSCLC cells, as well as tumor growth and metastasis in mouse models. Additionally, silencing of CTDSPL2 enhanced CD4+ T cell infiltration into tumors. Moreover, CTDSPL2 interacted with JAK1 and positively regulated JAK1 expression. Subsequent experiments indicated that CTDSPL2 activated the PI3K/AKT signaling pathway through the upregulation of JAK1, thereby promoting the progression of NSCLC. In conclusion, CTDSPL2 may play an oncogenic role in NSCLC progression by activating PI3K/AKT signaling via JAK1. These findings may provide a potential target for the diagnosis and treatment of NSCLC.
期刊介绍:
Cell Death Discovery is a multidisciplinary, international, online-only, open access journal, dedicated to publishing research at the intersection of medicine with biochemistry, pharmacology, immunology, cell biology and cell death, provided it is scientifically sound. The unrestricted access to research findings in Cell Death Discovery will foster a dynamic and highly productive dialogue between basic scientists and clinicians, as well as researchers in industry with a focus on cancer, neurobiology and inflammation research. As an official journal of the Cell Death Differentiation Association (ADMC), Cell Death Discovery will build upon the success of Cell Death & Differentiation and Cell Death & Disease in publishing important peer-reviewed original research, timely reviews and editorial commentary.
Cell Death Discovery is committed to increasing the reproducibility of research. To this end, in conjunction with its sister journals Cell Death & Differentiation and Cell Death & Disease, Cell Death Discovery provides a unique forum for scientists as well as clinicians and members of the pharmaceutical and biotechnical industry. It is committed to the rapid publication of high quality original papers that relate to these subjects, together with topical, usually solicited, reviews, editorial correspondence and occasional commentaries on controversial and scientifically informative issues.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


