Marina Tesi, Roberto Cammi, Giovanni Granucci, Maurizio Persico
{"title":"An algorithm for very high pressure molecular dynamics simulations","authors":"Marina Tesi, Roberto Cammi, Giovanni Granucci, Maurizio Persico","doi":"10.1002/jcc.27461","DOIUrl":null,"url":null,"abstract":"<p>We describe a method to run simulations of ground or excited state dynamics under extremely high pressures. The method is based on the introduction of a fictitious ideal gas that exerts the required pressure on a molecular sample and is therefore called XP-GAS (eXtreme Pressure by Gas Atoms in a Sphere). The algorithm is most suitable for approximately spherical clusters of molecules described by quantum chemistry methods, Molecular Mechanics or mixed QM/MM approaches. We compare the results obtained by the algorithm here presented and by the XP-PCM approach, based on a continuum description of the environment. As a test case, we study the conformational dynamics of 1,3-butadiene either as an isolated molecule (“naked” butadiene) or embedded in a cluster of argon atoms, under pressures up to 15 GPa. Overall, our results show that the XP-GAS QM/MM simulation method is in good agreement with the XP-PCM QM/Continuum model (Cammi model) in describing the effect of the pressure on static properties as the equilibrium geometry of butadiene in the ground state. Furthermore, the comparison of XP-GAS simulations with naked butadiene and butadiene in argon shows the importance, for XP-GAS and related methods, of a realistic representation of the medium in modelling pressure effects.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2848-2861"},"PeriodicalIF":3.4000,"publicationDate":"2024-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27461","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27461","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
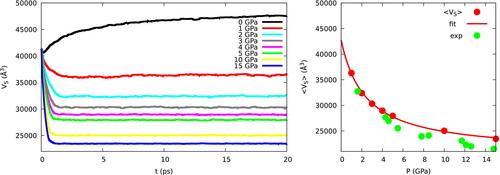
We describe a method to run simulations of ground or excited state dynamics under extremely high pressures. The method is based on the introduction of a fictitious ideal gas that exerts the required pressure on a molecular sample and is therefore called XP-GAS (eXtreme Pressure by Gas Atoms in a Sphere). The algorithm is most suitable for approximately spherical clusters of molecules described by quantum chemistry methods, Molecular Mechanics or mixed QM/MM approaches. We compare the results obtained by the algorithm here presented and by the XP-PCM approach, based on a continuum description of the environment. As a test case, we study the conformational dynamics of 1,3-butadiene either as an isolated molecule (“naked” butadiene) or embedded in a cluster of argon atoms, under pressures up to 15 GPa. Overall, our results show that the XP-GAS QM/MM simulation method is in good agreement with the XP-PCM QM/Continuum model (Cammi model) in describing the effect of the pressure on static properties as the equilibrium geometry of butadiene in the ground state. Furthermore, the comparison of XP-GAS simulations with naked butadiene and butadiene in argon shows the importance, for XP-GAS and related methods, of a realistic representation of the medium in modelling pressure effects.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


