A molecular mechanism for angiotensin II receptor blocker-mediated slit membrane protection: Angiotensin II increases nephrin endocytosis via AT1-receptor-dependent ERK 1/2 activation
Eva Königshausen, Ulf M. Zierhut, Martin Ruetze, Lars C. Rump, Lorenz Sellin
{"title":"A molecular mechanism for angiotensin II receptor blocker-mediated slit membrane protection: Angiotensin II increases nephrin endocytosis via AT1-receptor-dependent ERK 1/2 activation","authors":"Eva Königshausen, Ulf M. Zierhut, Martin Ruetze, Lars C. Rump, Lorenz Sellin","doi":"10.1096/fj.202400369R","DOIUrl":null,"url":null,"abstract":"<p>Albuminuria is characterized by a disruption of the glomerular filtration barrier, which is composed of the fenestrated endothelium, the glomerular basement membrane, and the slit diaphragm. Nephrin is a major component of the slit diaphragm. Apart from hemodynamic effects, Ang II enhances albuminuria by β-Arrestin2-mediated nephrin endocytosis. Blocking the AT1 receptor with candesartan and irbesartan reduces the Ang II-mediated nephrin-β-Arrestin2 interaction. The inhibition of MAPK ERK 1/2 blocks Ang II-enhanced nephrin-β-Arrestin2 binding. ERK 1/2 signaling, which follows AT1 receptor activation, is mediated by G-protein signaling, EGFR transactivation, and β-Arrestin2 recruitment. A mutant AT1 receptor defective in EGFR transactivation and β-Arrestin2 recruitment reduces the Ang II-mediated increase in nephrin β-Arrestin2 binding. The mutation of β-Arrestin2<sup>K11,K12</sup>, critical for AT1 receptor binding, completely abrogates the interaction with nephrin, independent of Ang II stimulation. β-Arrestin2<sup>K11R,K12R</sup> does not influence nephrin cell surface expression. The data presented here deepen our molecular understanding of a blood-pressure-independent molecular mechanism of AT-1 receptor blockers (ARBs) in reducing albuminuria.</p>","PeriodicalId":50455,"journal":{"name":"FASEB Journal","volume":null,"pages":null},"PeriodicalIF":4.4000,"publicationDate":"2024-08-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1096/fj.202400369R","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"FASEB Journal","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1096/fj.202400369R","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
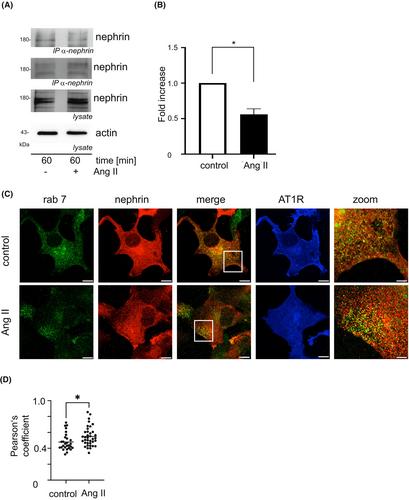
Albuminuria is characterized by a disruption of the glomerular filtration barrier, which is composed of the fenestrated endothelium, the glomerular basement membrane, and the slit diaphragm. Nephrin is a major component of the slit diaphragm. Apart from hemodynamic effects, Ang II enhances albuminuria by β-Arrestin2-mediated nephrin endocytosis. Blocking the AT1 receptor with candesartan and irbesartan reduces the Ang II-mediated nephrin-β-Arrestin2 interaction. The inhibition of MAPK ERK 1/2 blocks Ang II-enhanced nephrin-β-Arrestin2 binding. ERK 1/2 signaling, which follows AT1 receptor activation, is mediated by G-protein signaling, EGFR transactivation, and β-Arrestin2 recruitment. A mutant AT1 receptor defective in EGFR transactivation and β-Arrestin2 recruitment reduces the Ang II-mediated increase in nephrin β-Arrestin2 binding. The mutation of β-Arrestin2K11,K12, critical for AT1 receptor binding, completely abrogates the interaction with nephrin, independent of Ang II stimulation. β-Arrestin2K11R,K12R does not influence nephrin cell surface expression. The data presented here deepen our molecular understanding of a blood-pressure-independent molecular mechanism of AT-1 receptor blockers (ARBs) in reducing albuminuria.
血管紧张素 II 受体阻滞剂介导裂隙膜保护的分子机制:血管紧张素 II 通过 AT1 受体依赖性 ERK 1/2 激活增加肾素内吞
肾小球滤过屏障由栅栏状内皮、肾小球基底膜和裂隙隔膜组成,白蛋白尿的特征是肾小球滤过屏障遭到破坏。肾素是裂隙隔膜的主要组成部分。除了血流动力学效应外,Ang II 还会通过 β-Arrestin2 介导的肾素内吞作用增加白蛋白尿。用坎地沙坦和厄贝沙坦阻断 AT1 受体可减少 Ang II 介导的肾素-β-阿restin2 相互作用。抑制 MAPK ERK 1/2可阻止 Ang II 增强的肾素-β-阿瑞斯汀2结合。AT1 受体激活后的 ERK 1/2 信号传导是由 G 蛋白信号传导、表皮生长因子受体转录激活和 β-Arrestin2 招募介导的。在表皮生长因子受体反式激活和 β-Arrestin2 招募方面存在缺陷的突变 AT1 受体可减少 Ang II 介导的肾素 β-Arrestin2 结合的增加。对 AT1 受体结合至关重要的 β-Arrestin2K11,K12 基因突变会完全终止与肾素的相互作用,与 Ang II 的刺激无关。β-Arrestin2K11R,K12R 不影响肾素细胞表面的表达。本文提供的数据加深了我们对AT-1受体阻滞剂(ARBs)降低白蛋白尿的不依赖于血压的分子机制的理解。
期刊介绍:
The FASEB Journal publishes international, transdisciplinary research covering all fields of biology at every level of organization: atomic, molecular, cell, tissue, organ, organismic and population. While the journal strives to include research that cuts across the biological sciences, it also considers submissions that lie within one field, but may have implications for other fields as well. The journal seeks to publish basic and translational research, but also welcomes reports of pre-clinical and early clinical research. In addition to research, review, and hypothesis submissions, The FASEB Journal also seeks perspectives, commentaries, book reviews, and similar content related to the life sciences in its Up Front section.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


