{"title":"Phenotypic Spectrum and Natural History of Gillespie Syndrome. An Updated Literature Review with 2 New Cases.","authors":"Claudia Ciaccio, Matilde Taddei, Chiara Pantaleoni, Marina Grisoli, Daniela Di Bella, Stefania Magri, Franco Taroni, Stefano D'Arrigo","doi":"10.1007/s12311-024-01733-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Gillespie syndrome is a rare disorder caused by pathogenic variants in ITPR1 gene and characterized by the typical association of cerebellar ataxia, bilateral aniridia and intellectual disability. Since its first description in 1965, less than 100 patients have been reported and only 30 with a molecular confirmation.</p><p><strong>Methods: </strong>We present two additional cases, both carrying a loss-of-function variant in the Gly2539 amino acid residue. We describe the clinical evolution of the patients, one of whom is now 17 years old, and discuss the updated phenotypic spectrum of the disorder.</p><p><strong>Results: </strong>The study gives an overview on the condition, allowing to confirm important data, such as an overall positive evolution of development (with some patient not presenting intellectual disability), a clinical stability of the neurological signs (regardless of a possible progression of cerebellar atrophy) and ocular aspects, and a low prevalence of general health comorbidities.</p><p><strong>Discussion: </strong>Data about development and the observation of middle-aged patients lend support to the view that Gillespie is to be considered a non-progressive cerebellar ataxia, making this concept a key point for both clinicians and therapists, and for the families.</p>","PeriodicalId":50706,"journal":{"name":"Cerebellum","volume":null,"pages":null},"PeriodicalIF":2.7000,"publicationDate":"2024-08-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cerebellum","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s12311-024-01733-7","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Gillespie syndrome is a rare disorder caused by pathogenic variants in ITPR1 gene and characterized by the typical association of cerebellar ataxia, bilateral aniridia and intellectual disability. Since its first description in 1965, less than 100 patients have been reported and only 30 with a molecular confirmation.
Methods: We present two additional cases, both carrying a loss-of-function variant in the Gly2539 amino acid residue. We describe the clinical evolution of the patients, one of whom is now 17 years old, and discuss the updated phenotypic spectrum of the disorder.
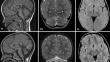
Results: The study gives an overview on the condition, allowing to confirm important data, such as an overall positive evolution of development (with some patient not presenting intellectual disability), a clinical stability of the neurological signs (regardless of a possible progression of cerebellar atrophy) and ocular aspects, and a low prevalence of general health comorbidities.
Discussion: Data about development and the observation of middle-aged patients lend support to the view that Gillespie is to be considered a non-progressive cerebellar ataxia, making this concept a key point for both clinicians and therapists, and for the families.
期刊介绍:
Official publication of the Society for Research on the Cerebellum devoted to genetics of cerebellar ataxias, role of cerebellum in motor control and cognitive function, and amid an ageing population, diseases associated with cerebellar dysfunction.
The Cerebellum is a central source for the latest developments in fundamental neurosciences including molecular and cellular biology; behavioural neurosciences and neurochemistry; genetics; fundamental and clinical neurophysiology; neurology and neuropathology; cognition and neuroimaging.
The Cerebellum benefits neuroscientists in molecular and cellular biology; neurophysiologists; researchers in neurotransmission; neurologists; radiologists; paediatricians; neuropsychologists; students of neurology and psychiatry and others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


