Miaomiao Qiu, Xiaoming Zhao, Tao Guo, Hongyun He, Yihao Deng
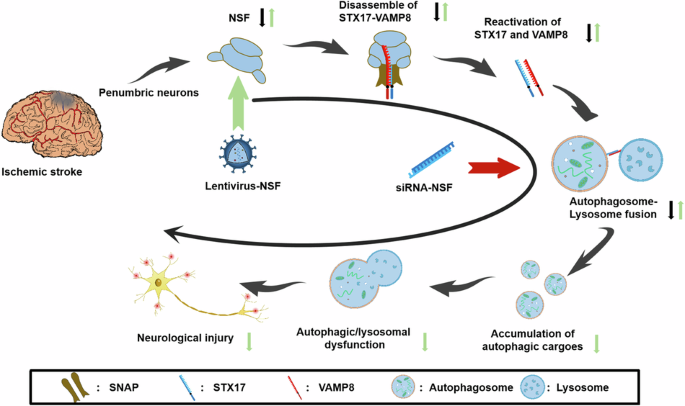
{"title":"N-ethylmaleimide-sensitive factor elicits a neuroprotection against ischemic neuronal injury by restoring autophagic/lysosomal dysfunction.","authors":"Miaomiao Qiu, Xiaoming Zhao, Tao Guo, Hongyun He, Yihao Deng","doi":"10.1038/s41420-024-02144-7","DOIUrl":null,"url":null,"abstract":"<p><p>Autophagosome-lysosome fusion defects play a critical role in driving autolysosomal dysfunction, leading to autophagic/lysosomal impairment in neurons following ischemic stroke. However, the mechanisms hindering autophagosome-lysosome fusion remain unclear. Soluble N-ethylmaleimide-sensitive factor (NSF) is an essential ATPase to reactivate STX17 and VAMP8, which are the paired molecules to mediate fusion between autophagosomes and lysosomes. However, NSF is frequently inactivated to inhibit the reactivation of STX17 and VAMP8 in ischemic neurons. Herein, we investigated whether autophagosome-lysosome fusion could be facilitated to alleviate autophagic/lysosomal impairment in ischemic neurons by over-expressing NSF. Rat model of middle cerebral artery occlusion (MCAO) and HT22 neuron ischemia model of oxygen-glucose deprivation (OGD) were prepared, respectively. The results demonstrated that NSF activity was significantly suppressed, accompanied by reduced expressions of STX17 and VAMP8 in penumbral neurons 48 h post-MCAO and in HT22 neurons 2 h post-OGD. Moreover, the attenuated autolysosome formation accompanied by autophagic/lysosomal dysfunction was observed. Thereafter, NSF activity in HT22 neurons was altered by over-expression and siRNA knockdown, respectively. After transfection with recombinant NSF-overexpressing lentiviruses, both STX17 and VAMP8 expressions were concurrently elevated to boost autophagosome-lysosome fusion, as shown by enhanced immunofluorescence intensity co-staining with LC3 and LAMP-1. Consequently, the OGD-created autophagic/lysosomal dysfunction was prominently ameliorated, as reflected by augmented autolysosomal functions and decreased autophagic substrates. By contrast, NSF knockdown conversely aggravated the autophagic/lysosomal impairment, and thereby exacerbated neurological damage. Our study indicates that NSF over-expression induces neuroprotection against ischemic neuronal injury by restoring autophagic/lysosomal dysfunction via the facilitation of autophagosome-lysosome fusion. Over-expression of NSF promotes fusion by reactivating STX17 and VAMP8. Black arrows represent the pathological process after cerebral ischemia, green arrows represent the mechanism of remission after NSF over-expression, and red arrows represent the effect on the pathological process after NSF knockdown.</p>","PeriodicalId":9735,"journal":{"name":"Cell Death Discovery","volume":null,"pages":null},"PeriodicalIF":6.1000,"publicationDate":"2024-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11330971/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Death Discovery","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41420-024-02144-7","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Autophagosome-lysosome fusion defects play a critical role in driving autolysosomal dysfunction, leading to autophagic/lysosomal impairment in neurons following ischemic stroke. However, the mechanisms hindering autophagosome-lysosome fusion remain unclear. Soluble N-ethylmaleimide-sensitive factor (NSF) is an essential ATPase to reactivate STX17 and VAMP8, which are the paired molecules to mediate fusion between autophagosomes and lysosomes. However, NSF is frequently inactivated to inhibit the reactivation of STX17 and VAMP8 in ischemic neurons. Herein, we investigated whether autophagosome-lysosome fusion could be facilitated to alleviate autophagic/lysosomal impairment in ischemic neurons by over-expressing NSF. Rat model of middle cerebral artery occlusion (MCAO) and HT22 neuron ischemia model of oxygen-glucose deprivation (OGD) were prepared, respectively. The results demonstrated that NSF activity was significantly suppressed, accompanied by reduced expressions of STX17 and VAMP8 in penumbral neurons 48 h post-MCAO and in HT22 neurons 2 h post-OGD. Moreover, the attenuated autolysosome formation accompanied by autophagic/lysosomal dysfunction was observed. Thereafter, NSF activity in HT22 neurons was altered by over-expression and siRNA knockdown, respectively. After transfection with recombinant NSF-overexpressing lentiviruses, both STX17 and VAMP8 expressions were concurrently elevated to boost autophagosome-lysosome fusion, as shown by enhanced immunofluorescence intensity co-staining with LC3 and LAMP-1. Consequently, the OGD-created autophagic/lysosomal dysfunction was prominently ameliorated, as reflected by augmented autolysosomal functions and decreased autophagic substrates. By contrast, NSF knockdown conversely aggravated the autophagic/lysosomal impairment, and thereby exacerbated neurological damage. Our study indicates that NSF over-expression induces neuroprotection against ischemic neuronal injury by restoring autophagic/lysosomal dysfunction via the facilitation of autophagosome-lysosome fusion. Over-expression of NSF promotes fusion by reactivating STX17 and VAMP8. Black arrows represent the pathological process after cerebral ischemia, green arrows represent the mechanism of remission after NSF over-expression, and red arrows represent the effect on the pathological process after NSF knockdown.
期刊介绍:
Cell Death Discovery is a multidisciplinary, international, online-only, open access journal, dedicated to publishing research at the intersection of medicine with biochemistry, pharmacology, immunology, cell biology and cell death, provided it is scientifically sound. The unrestricted access to research findings in Cell Death Discovery will foster a dynamic and highly productive dialogue between basic scientists and clinicians, as well as researchers in industry with a focus on cancer, neurobiology and inflammation research. As an official journal of the Cell Death Differentiation Association (ADMC), Cell Death Discovery will build upon the success of Cell Death & Differentiation and Cell Death & Disease in publishing important peer-reviewed original research, timely reviews and editorial commentary.
Cell Death Discovery is committed to increasing the reproducibility of research. To this end, in conjunction with its sister journals Cell Death & Differentiation and Cell Death & Disease, Cell Death Discovery provides a unique forum for scientists as well as clinicians and members of the pharmaceutical and biotechnical industry. It is committed to the rapid publication of high quality original papers that relate to these subjects, together with topical, usually solicited, reviews, editorial correspondence and occasional commentaries on controversial and scientifically informative issues.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


