{"title":"Difenoconazole solubility in acetonitrile/N,N-dimethylformamide/acetone + water and quantum chemistry study into inter/intra-molecular interactions","authors":"Chaobin Ren , Hongkun Zhao","doi":"10.1016/j.jct.2024.107366","DOIUrl":null,"url":null,"abstract":"<div><p>The difenoconazole solubilities in acetonitrile/acetone/<em>N</em>,<em>N</em>-dimethylformamide (DMF) + water systems were acquired experimentally with the help of the isothermal shake-flask technique. Analysis of X-ray power diffraction revealed that difenoconazole did not exhibit any crystal transition as well as solvate formation. The solubility acquired here was accurately correlated, which yielded relative average deviations (<em>RAD</em>) of ≤4.99 % and a root-mean-square deviation of ≤20.59 × 10<sup>−5</sup> through the Jouyban-Acree and modified van’t Hoff-Jouyban-Acree models Additionally, to explain the solubility behavior at a temperature of 298.15 K, this work examines the acetonitrile/acetone/DMF + water and the previously published methanol/ethanol/isopropanol/PG + water blends utilizing the extended Hildebrand solubility technique. The <em>RAD</em> levels remained below 6.88 %. The dipolarity-polarizability and the solubility parameter of blended solvents exert a substantial impact on the variability of difenoconazole solubility. The preferred solvation of difenoconazole at a temperature of 298.15 K was examined through the inverse Kirkwood-Buff integrals. The solvation parameters’ values of difenoconazole were positive in blends containing methanol, acetone, DMF, ethanol, isopropanol, and acetonitrile with moderate and rich compositions. This indicates that difenoconazole is preferentially solvated by them in above composition ranges. A shift from an enthalpy-driven mechanism to an entropy-driven one was revealed through an analysis of the thermodynamics of the entropy-enthalpy relationship in the dissolution of difenoconazole in blends. Furthermore, the mean local ionization energy, Hirshfeld surface as well as molecular surface electrostatic potential were employed to illustrate the microscopic electrostatic characteristics. The <img>N<img> and <img>N<img> groups in the five-membered ring of difenoconazole molecule are the primary sites for electrophilic attack. The weak contacts of difenoconazole-solvent were demonstrated with the help of a Hirshfeld partition analysis-based independent gradient model.</p></div>","PeriodicalId":54867,"journal":{"name":"Journal of Chemical Thermodynamics","volume":"200 ","pages":"Article 107366"},"PeriodicalIF":2.2000,"publicationDate":"2024-08-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Thermodynamics","FirstCategoryId":"5","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0021961424001198","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
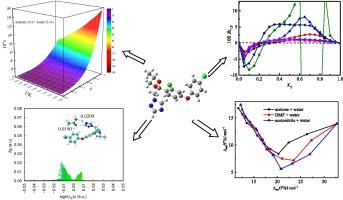
Abstract
The difenoconazole solubilities in acetonitrile/acetone/N,N-dimethylformamide (DMF) + water systems were acquired experimentally with the help of the isothermal shake-flask technique. Analysis of X-ray power diffraction revealed that difenoconazole did not exhibit any crystal transition as well as solvate formation. The solubility acquired here was accurately correlated, which yielded relative average deviations (RAD) of ≤4.99 % and a root-mean-square deviation of ≤20.59 × 10−5 through the Jouyban-Acree and modified van’t Hoff-Jouyban-Acree models Additionally, to explain the solubility behavior at a temperature of 298.15 K, this work examines the acetonitrile/acetone/DMF + water and the previously published methanol/ethanol/isopropanol/PG + water blends utilizing the extended Hildebrand solubility technique. The RAD levels remained below 6.88 %. The dipolarity-polarizability and the solubility parameter of blended solvents exert a substantial impact on the variability of difenoconazole solubility. The preferred solvation of difenoconazole at a temperature of 298.15 K was examined through the inverse Kirkwood-Buff integrals. The solvation parameters’ values of difenoconazole were positive in blends containing methanol, acetone, DMF, ethanol, isopropanol, and acetonitrile with moderate and rich compositions. This indicates that difenoconazole is preferentially solvated by them in above composition ranges. A shift from an enthalpy-driven mechanism to an entropy-driven one was revealed through an analysis of the thermodynamics of the entropy-enthalpy relationship in the dissolution of difenoconazole in blends. Furthermore, the mean local ionization energy, Hirshfeld surface as well as molecular surface electrostatic potential were employed to illustrate the microscopic electrostatic characteristics. The N and N groups in the five-membered ring of difenoconazole molecule are the primary sites for electrophilic attack. The weak contacts of difenoconazole-solvent were demonstrated with the help of a Hirshfeld partition analysis-based independent gradient model.
期刊介绍:
The Journal of Chemical Thermodynamics exists primarily for dissemination of significant new knowledge in experimental equilibrium thermodynamics and transport properties of chemical systems. The defining attributes of The Journal are the quality and relevance of the papers published.
The Journal publishes work relating to gases, liquids, solids, polymers, mixtures, solutions and interfaces. Studies on systems with variability, such as biological or bio-based materials, gas hydrates, among others, will also be considered provided these are well characterized and reproducible where possible. Experimental methods should be described in sufficient detail to allow critical assessment of the accuracy claimed.
Authors are encouraged to provide physical or chemical interpretations of the results. Articles can contain modelling sections providing representations of data or molecular insights into the properties or transformations studied. Theoretical papers on chemical thermodynamics using molecular theory or modelling are also considered.
The Journal welcomes review articles in the field of chemical thermodynamics but prospective authors should first consult one of the Editors concerning the suitability of the proposed review.
Contributions of a routine nature or reporting on uncharacterised materials are not accepted.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


